















- DAZ.online
- DAZ / AZ
- DAZ 15/2009
- Individuelle ...
Interpharm 2009
Individuelle Gendiagnostik – Pro und Contra
Dr. Ilse Zündorf
Wie sehr wir im "Gen-Zeitalter" angekommen sind, haben wahrscheinlich viele noch gar nicht begriffen. Ein vorläufiger Höhepunkt war die Publikation der ersten Draft-Version des kompletten humanen Genoms im Februar 2001, der 2003 die finale Version folgte. Zwischenzeitlich ist eine fast unüberschaubare Zahl aller möglicher Genome sequenziert, darunter 842 mikrobielle und 531 eukaryontische Genome. Von den eukaryontischen Genomen sind derzeit 23 komplett abgeschlossen. Bei 251 werden die Daten gerade zusammengeführt, und 257 Genome werden fortgeschritten bearbeitet (www.ncbi.nlm.nih.gov/genomes/leuks.cgi). Und auch das nächste Ziel ist bereits definiert: Das 1000 Genome Project, ein internationales Projekt, das im Januar 2008 initiiert wurde und dessen Ziel es ist, in den folgenden drei Jahren die Genome von rund 1000 Menschen vollständig zu sequenzieren, um daraus einen höchst detaillierten Katalog menschlicher genetischer Variationen zu erstellen (www.1000genomes.org/page.php).
Diese enorme Masse an genomischer Information darf aber nicht darüber hinwegtäuschen, dass wir noch weit davon entfernt sind, Genome auch zu verstehen. Die große Herausforderung innerhalb der Aufklärung von Genomen bildet die Assoziation von Sequenzen mit Funktionen und gleichermaßen die Assoziation von Sequenzvariationen mit Funktionsstörungen.
Nichtsdestotrotz ist es heute bereits möglich, auf eigene genomische Daten zuzugreifen. Firmen wie 23andme, deCODE Genetics, DNAdirect, Knome, Navigenics u. a. bieten heute für akzeptables Geld jedem Zahlungswilligen an, einen Blick in das eigene Genom zu werfen. Was man mit solchen Daten anfangen kann, ist äußerst umstritten, und schließlich muss jeder selbst entscheiden, ob er einen entsprechenden Auftrag unterschreibt. Wer sich jedoch zu einem solchen Schritt entschließt und vielleicht bereit ist, mehr zu erfahren, als das, was man von den Firmen geliefert bekommt, dem erschließen sich gewaltige Möglichkeiten.
Biologische Grundlagen
Dank des humanen Genomprojekts kennt man heute die exakte Zahl an Nukleotiden, die ein menschliches Genom enthält: Etwa 3,2 Milliarden Basenpaare codieren für ein haploides Genom und etwa 6,5 Milliarden Basenpaare für ein diploides Genom. Diese Basenpaare verteilen sich beim Menschen auf 46 Chromosomen – 44 Autosomen und die beiden Gonosomen X und Y.
Punktmutationen, also Veränderungen an einer einzelnen Stelle in der DNA, treten im Schnitt bei jeder 1000. Base auf. Man nennt diese Punktmutationen SNPs (single nucleotide polymorphisms). Das bedeutet, dass sich zwei Genome näherungsweise in ca. 6,5 Millionen Bausteinen unterscheiden können. Umgekehrt sind ca. 5,9 Milliarden Bausteine bei zwei Genomen identisch.
Prof. Dr. Theo Dingermann
Das HapMap-Projekt, das als eines der Nachfolgeprojekte des Human Genome Project initiiert wurde, ergab, dass in allen humanen Genomen ca. 12 Millionen Basenpositionen variabel sein können. Nahezu alle diese 12 Millionen SNPs wurden im Rahmen des HapMap-Projekts zwischenzeitlich identifiziert und charakterisiert. Ein weiteres Resultat dieses Projektes war die Erkenntnis, dass SNPs in bestimmten Mustern, den Haplotypen, vorkommen. Das bedeutet, dass von einzelnen SNPs auf andere SNPs in der Nachbarschaft geschlossen werden kann.
Ein Großteil dieser Variabilität ist entweder "ganz normal", unschädlich oder gar von Vorteil. Die "ganz normale" Variabilität befindet sich in Genombereichen, die dafür verantwortlich sind, dass sich Individuen voneinander unterscheiden – beispielsweise in ihrem Aussehen.
Andere Veränderungen sind unschädlich, weil nicht alle Bereiche des Genoms als "informative Bereiche" ausgelegt sind. Jede Mutation in den nicht-informativen Bereichen ist biologisch neutral.
Selten sind Mutationen sogar potenziell von Vorteil. So sind Menschen mit einem mutierten Gen für Thalassämien (inklusive der Sichelzellanämie) oder mit einer Glucose-6-phosphatdehydrogenase-Defizienz besser vor Malariaerregern geschützt. Es sind auch Mutationen bekannt, die sehr effektiv vor einer HIV-Infektion schützen.
Somit muss nur ein relativ kleiner Rest der variablen Positionen als pathologisch oder potenziell pathologisch angesehen werden. Selbst wenn eine Mutation in einem Gen auftritt, kann der Defekt vielfach durch eine intakte Kopie dieses Gens (Allel) auf dem Partnerchromosom kompensiert werden. Allerdings erhöhen derartige Mutationen das Risiko, zu erkranken – nämlich dann, wenn zusätzlich auch das (noch) intakte Allel mutiert.
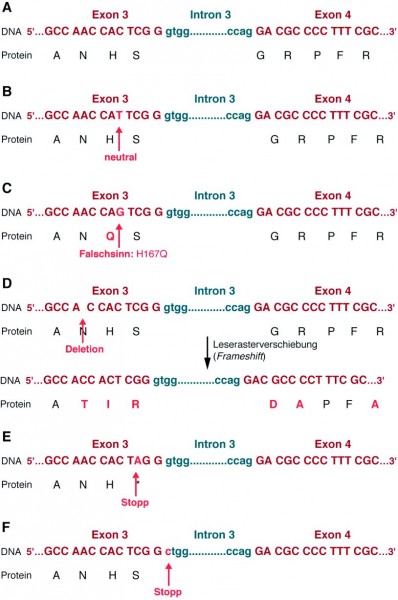
Abb. 1: Mögliche Konsequenzen von SNPs.
A. Ausschnitt einer DNA-Sequenz eines Proteins mit den beiden Protein-codierenden Exons 3 und 4 und dem dazwischen liegenden nicht-codierenden Intron. Diese Sequenz sei die am häufigsten in der Population vorkommende DNA-Sequenz, also die "Wildtyp"-Sequenz.
B. Die C → T-Mutation ändert ein Codon von CAC zu CAT. Da beide Codons für die Aminosäure Histidin stehen, ändert sich die Primärstruktur des Proteins nicht. Es handelt sich um eine "stille" Mutation.
C. Eine C → G-Mutation ändert das Codon CAC zu CAG und damit die Proteinsequenz an dieser Stelle von Histidin zu Glutamin. Dies ist eine Falschsinn-(Missense) -Mutation, die gegebenenfalls die Aktivität des Enzyms verändern kann.
D. Wenn eine oder auch zwei Basen verlorengehen bzw. hinzukommen, ändert sich der Leserahmen (die Triplettfolge), sodass eine andere Aminosäurensequenz codiert wird. Oft folgt in dem falschen Leserahmen sehr bald ein Stopp-Codon, was zu einem verkürzten und damit funktionslosen Protein führt.
E. Eine Mutation kann auch direkt ein Stopp-Codon generieren. Auch in diesem Fall entsteht ein verkürztes, funktionsloses Protein.
F. Selbst wenn eine Mutation nicht im Exon, sondern im Intronbereich vorkommt, kann das fatale Folgen haben. Im gezeigten Beispiel wurde durch eine G → C-Mutation die Spleiß-Donor-Sequenz zerstört. Als Folge wird die mRNA des Gens nicht mehr korrekt prozessiert. Da das Intron nun nicht mehr entfernt wird, unterbricht die Intronsequenz den Leserahmen zwischen den Exons 3 und 4, was zu einem defekten Protein führt.
Unterschiedliche "Qualität" von Punktmutationen
Nicht alle Punktmutationen führen zu biologischen Konsequenzen, da sie beispielsweise das Codierungsverhalten nicht ändern. In solchen Fällen werden Basen ausgetauscht, die zwar die Codonstruktur ändern, aber so, dass das neu entstandene Codon für die gleiche Aminosäure kodiert wie das ursprüngliche Codon (Abb. 1B). Dieser Mutations-Typ auch als "stille Mutation" oder als einen "synonymen Basenaustausch" bezeichnet. "Stille Mutationen" in Genen lassen sich daher nur auf DNA-Ebene und nicht auf Protein-Ebene erkennen.
Unter einer Missense-Mutation versteht man einen Basenaustausch, der zur Codierung einer anderen Aminosäure führt (Abb. 1C). Dies kann, muss sich aber nicht auf die Funktion des jeweiligen Proteins auswirken, je nachdem an welcher Stelle im Protein sich die Variation befindet und je nachdem ob sich die ursprüngliche und die neue Aminosäure in ihrem chemischen Charakter stark unterscheiden bzw. eher ähnlich sind.
Besonders gefürchtet sind Nonsense- und Leseraster (Frameshift)-Mutationen. Bei den Nonsense-Mutationen wird ein Codon für eine Aminosäure in ein Stopp-Codon umgewandelt. Dies führt zur vorzeitigen Termination der Proteinbiosynthese, sodass verkürzte Proteinvariationen resultieren, die in aller Regel funktionslos sind (Abb. 1E). Etliche monogenetische Erbkrankheiten lassen sich auf Nonsense-Mutationen in einem relevanten Gen zurückführen. Zum Beispiel führt die Nonsense-Mutationen NS 39 CAG → TAG (die Umwandlung des ersten Nukleotids des Codons 39 von C nach T) zu einer schweren Form der β-Thalassämie.
Bei Leseraster (Frameshift)-Mutationen geht in aller Regel eine Base verloren, oder es wird im Laufe der Replikation eine Base zu viel eingefügt (Abb. 1D). Dies ist – ähnlich wie die Nonsense-Mutation – ein sehr schwerwiegendes Ereignis, da sich dadurch das ursprüngliche Dreierraster der Aminosäurencodierung verschiebt. Es entstehen im Anschluss an die Mutationsposition völlig neue Proteinsequenzen, die absolut keinen Sinn machen, sodass die resultierenden Proteine auch in aller Regel funktionslos sind.
Väterliche und mütterliche Abstammung
Während der Meiose kommt es zum crossing-over homologer Chromosomen väterlichen und mütterlichen Ursprungs. Aus diesem Grund kann man nicht ein bestimmtes Chromosom eines Kindes entweder dem Vater oder der Mutter zuordnen. Allerdings gibt es zwei Ausnahmen, die es erlauben, aufgrund genetischer Daten die väterliche (paternale) bzw. die mütterliche (maternale) Abstammung zurückzuverfolgen.
Ein väterlicher Stammbaum kann auf der Basis des Y-Chromosoms erstellt werden. Da es nur ein Y-Chromosom gibt, kann es nicht zu einem crossing-over kommen, sodass die Sequenzen des Y-Chromosoms komplett und unverändert an die nächste (männliche) Generation weitergegeben werden.
Eine Abstammung mütterlicherseits kann über das mitochondriale Genom rekonstruiert werden. Denn Mitochondrien werden ausschließlich von der Mutter an die nächste Generation beider Geschlechter weitergegeben.
Kommerzielle individuelle Genom-Analyse
Entschließt man sich, individuelle genomische Information zu "kaufen", stellt man sich sicherlich mehrere Fragen, wie z. B.:
- Was kostet eine Analyse?
- Wie werden die Daten ermittelt?
- Wie werden die Daten übermittelt und werden sie interpretiert?
- Bekommt man auch Zugriff auf die Primärdaten und kann man diese auch selber weiter auswerten?
Diese Fragen müssen sicherlich unterschiedlich beantwortet werden, je nachdem welche Firma man mit der Analyse beauftragt.
Dingermann und Zündorf haben ihre genomischen Sequenzinformationen bei der kalifornischen Firma 23andme (www.23andme.com) in Auftrag gegeben. Hier kostet derzeit die Analyse 399 US-Dollar. Dieser Preis ist erstaunlich niedrig. Beliefen sich die Kosten für die im humanen Genomprojekt ermittelten Sequenz noch auf 300 Millionen US-Dollar, kann man mit der aktuell verfügbaren Sequenziertechnologie (Next-Generation-System) eine humane Genomsequenz mit einem Kostenaufwand von 60.000 US-Dollar erstellen. Wie kann es da sein, dass 23andme nur 399 US-Dollar verlangt?
Welche Augenfarbe? Welche Vorlieben?
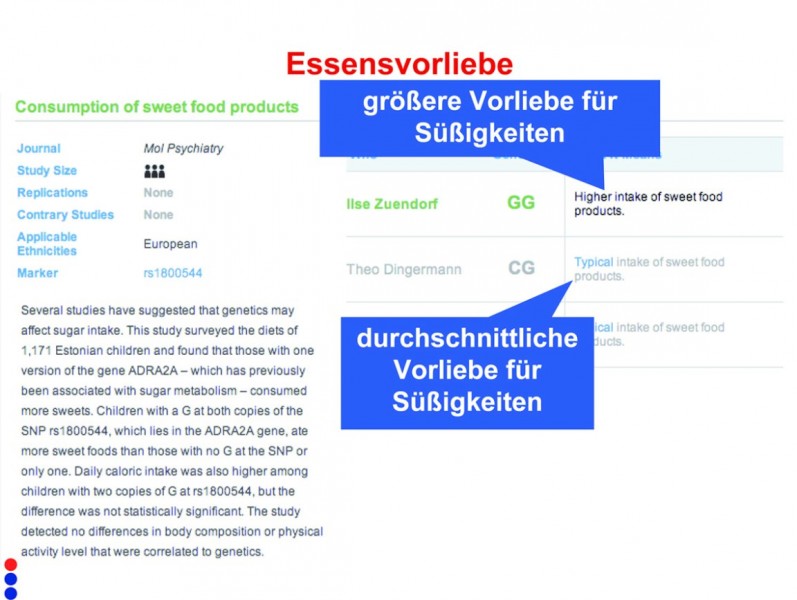
Eine kommerzielle individuelle Genom-Analyse ermöglicht es, aus den genomischen Sequenzinformationen Rückschlüsse auf sichtbare Eigenschaften, aber auch auf "unsichtbare" persönliche Vorlieben einer Person zu ziehen: So ergibt sich aus der Analyse des Genoms von Professor Dingermann (links), dass er zu 56% braune Augen hat, was deutlich zu sehen ist. Die Genom-Analyse von Frau Zündorf (rechts) bezüglich der Essensvorlieben verrät schon mehr: eine "größere Vorliebe für Süßigkeiten".
Zum einen liegt dies daran, dass bei Firmen wie 23andme nicht konventionell sequenziert wird. Vielmehr werden die Sequenzdaten mithilfe von DNA-Chips ermittelt. Dazu wird DNA, die aus einer Sputum-Probe isoliert wurde, zunächst in vitro amplifiziert, dann zu kürzeren Fragmenten degradiert und schließlich mit einem DNA-Chip hybridisiert, auf dem seinerseits ganz bestimmte DNA-Fragmente fixiert sind. Hier erfolgt nun der enzymatische Einbau eines einzelnen farblich markierten Nukleotids. Anhand des eingebauten Farbmarkers kann man rückschließen, welche Base an einer bestimmten SNP-Position in der genomischen DNA der Probanden-DNA vorhanden ist.
Die Cleverness dieses Ansatzes besteht darin, dass man sich nur den ca. zwölf Millionen variablen Positionen widmet und die ca. 5,9 Milliarden konstanten Positionen einfach ignoriert; denn diese kennt man ohnehin, weil sie in allen menschlichen Genomen gleich sind.
Aber selbst die zwölf Millionen variablen Positionen müssen nicht komplett abgefragt werden, da mithilfe weniger "Testermutationen" (Tag-SNPs) auf bekannte Haplotypen – also benachbarte Mutationsmuster – rückgeschlossen werden kann.
Die Firma 23andme ermittelt ca. 600.000 SNPs, was eine recht gute Abdeckung aller potenzieller Mutationen bedeuten kann.
Diese Daten werden tatsächlich als Primärdaten dem Auftraggeber zur Verfügung gestellt, sodass man auch die Primärdaten analysieren kann, wenn man sich ein wenig auskennt.
Übermittelt werden die Daten über eine bestimmte Homepage, die für jeden Kunden angelegt wird. Zwar kann man die Daten auch auf den eigenen Computer laden. Sie bleiben aber auch immer elektronisch zugänglich, was vielleicht dem einen oder anderen missfallen könnte.
Interpretationen werden derzeit in Form von 27 clinical reports und 78 research reports zur Verfügung gestellt. Das mag für ca. 600.000 Primärdaten relativ wenig erscheinen. Hier deutet sich an, wie relativ wenig man derzeit noch über Assoziationen von Mutationen mit biologischen Eigenschaften weiß. Allerdings steigt das Wissen rasant, und es ist ein besonderer Service des Unternehmens, dass dieses Wissen nach und nach auf die persönlichen Daten ohne Mehrkosten übertragen wird.
Schneller Metabolisierer?
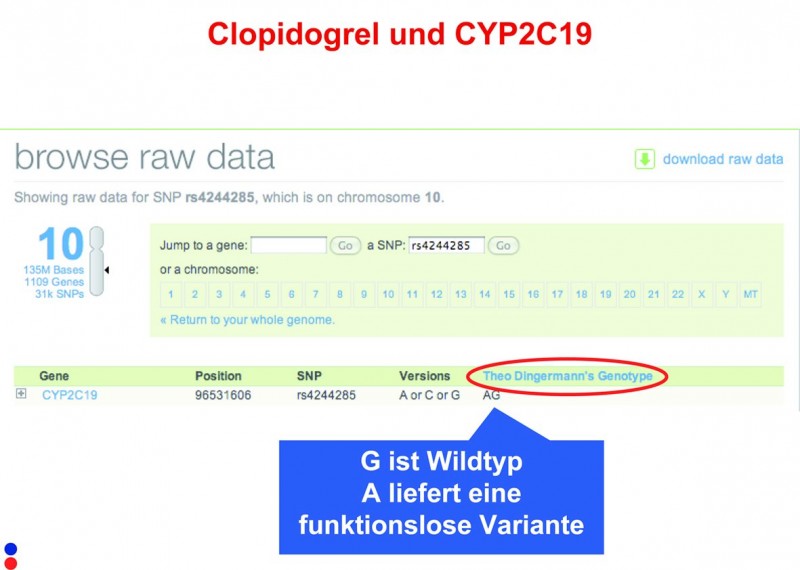
Interessanter als die Augenfarbe sind die Veranlagungen zur Verstoffwechslung von Arzneistoffen wie Coffein oder Statinen. Die Genom-Analyse zeigt, warum Frau Zündorf abends nach einer Tasse Kaffee nicht schlafen kann: Sie ist ein langsamer Coffein-Metabolisierer (rechts). Von größerer Relevanz ist das Wissen, ob man das entsprechende CYP-Enzym in ausreichender Aktivität besitzt, das z. B. Clopidogrel metabolisiert (links). Auch diese Informationen lassen sich dem Genom entnehmen – und möglicherweise missbrauchen.
Wer sich auskennt, kann aber auch die Primärdaten zurate ziehen, wenn neue Assoziationen publiziert werden. Beispielsweise wurde zu Beginn des Jahres im New England Journal of Medicine ein SNP identifiziert, der Aussagen darüber zulässt, ob ein Patient von der Gabe von Clopidogrel profitiert. Jeder SNP besitzt eine eindeutige Identifikationsnummer. Sucht man diese Nummer in seinen eigenen Daten, erhält man unmittelbar diese im Ernstfall doch nicht unerhebliche Information.
Individuelle Gendiagnostik Pro und Contra
Der Einblick in sein eigenes Genom geht weit über das hinaus, was man sich vielleicht als Laie unter dem Begriff "Gendiagnostik" vorstellt, denn die Diagnostik – zumindest die Diagnostik von Krankheiten – spielt eine deutlich untergeordnete Rolle.
In aller Regel werden "Risiken" offengelegt, nämlich dann, wenn ein Allel in der funktionellen Form und das zweite Allel in einer mutierten Form vorliegt. Dabei werden Krankheitsrisiken, die durch die Beteiligung mehrerer Gene beeinflusst werden, auch auf der Basis mehrerer SNPs beurteilt – natürlich nur so weit, wie es der aktuelle Wissensstand zulässt.
Es werden auch Eigenschaften bestimmt, die man in aller Regel bereits kennt, darunter die Augenfarbe, der Muskeltyp, der darüber entscheiden kann, ob man seine Veranlagung besser als Sprinter oder als Ausdauersportler nutzen sollte, die Fähigkeit "bitter" zu schmecken oder die Konsistenz des Ohrenschmalzes. Diese Informationen vermitteln nicht selten einen "Aha-Effekt" und sind zudem geeignet, die Zuverlässigkeit der Daten abzuschätzen.
Unter den research reports findet man Hinweise auf teils "kuriose", aber auch seriöse Risiken, die noch einer weiteren wissenschaftlichen Erhärtung bedürfen. Beispiele sind die genetische Anlage zum "Vermeiden von Fehlern", zur Ausprägung des Gedächtnisses, zur Veranlagung hinsichtlich einer Nicotinabhängigkeit usw., aber auch zum Brustkrebsrisiko, zur männlichen Infertilität oder zur Neigung zu Übergewicht.
Schließlich findet man Interessantes zur eigenen Abstammung heraus, indem man als Mann sowohl einen paternalen als auch einen maternalen Stammbaum, als Frau nur einen maternalen Stammbaum erstellen kann. Für Apothekerinnen und Apotheker besonders interessant sind die Veranlagungen zum physiologischen Umgang mit Arzneimitteln, wie Coffein, Statinen, β-Blockern usw. Hier bieten auch die Primärdaten einen unerschöpflichen Fundus, wenn man bereit ist, sich tiefer in die Materie einzuarbeiten.
Ob man all das wissen will oder ob man sich damit begnügt, zu wissen, dass man all das wissen könnte, bleibt letztlich jedem selbst überlassen.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.