











- DAZ.online
- DAZ / AZ
- DAZ 15/2008
- Schmerzmittel in der ...
Pharmakologie
Schmerzmittel in der Selbstmedikation
In diesem Beitrag wird eine – naturgemäß subjektive – (Neu-) Bewertung aller relevanten Wirkstoffe im rezeptfreien Verkauf (OTC) versucht. Dabei wird sich zeigen, dass Diclofenac und Ibuprofen zwar keineswegs ideale, aber im Vergleich die risikoärmeren Wirkstoffe sind. Das Propyphenazon könnte interessante Eigenschaften haben, ist allerdings nicht gründlich untersucht, sodass die möglichen Vorteile (zuverlässige Wirkung bei guter (?) Verträglichkeit) epidemiologisch unbelegt bleiben. Ähnliches gilt auch für das Phenazon, das zusätzlich mit zahlreichen Arzneimittelinteraktionen und bei einigen Patienten mit einer sehr langsamen Elimination behaftet ist. ASS, Paracetamol und Phenazon weisen erhebliche Risiken auf und können zur Therapie passagerer Schmerzen nicht mehr als erste Wahl gelten.
Wirkungsmechanismus: Hemmung der Cyclooxygenasen
Jahrzehntelang bestand Unklarheit darüber, wie diese Wirkstoffe ihre antipyretische, analgetische und zum Teil auch in therapeutischer Dosierung antiphlogistische Wirkung entfalten. Diese Frage ist heute beantwortet. Wir wissen, dass alle genannten Pharmaka die Cyclooxygenasen hemmen und damit ihren therapeutischen Effekt über eine Verminderung der Bildung der proalgetischen, proinflammatorischen und fieberauslösenden Prostaglandine erzielen [14 –16].
Die gewebeprotektiven und proalgetischen Prostaglandine können grundsätzlich von zwei unterschiedlichen Enzymen produziert werden: der Cyclooxygenase-1 (COX-1), die fast überall im Organismus vorhanden ist und im geringen Umfang gewebeprotektive Prostaglandine zur Verfügung stellt, und der Cyclooxygenase-2 (COX-2), die bei Entzündungen, Schmerzen, Infektionen usw. quasi als Adaptationsreaktion in bestimmten Organen gebildet wird [10]. So konnte kürzlich gezeigt werden, dass die COX-2 bei einer bakteriellen Infektion nicht nur am Ort der Infektion, sondern mithilfe von Zytokinen auch im Gehirn, im Gastrointestinaltrakt, in der Niere und in den Sexualorganen vermehrt gebildet wird [18].
Ganz ohne Prostaglandinproduktion ist menschliches und tierisches Leben unmöglich [27]. Das Fehlen der COX-1 oder COX-2 kann Probleme aufwerfen. Dieses gilt auch für die temporäre Blockade dieser Enzyme. Dementsprechend ist es von Bedeutung, dass in möglichst wenigen Organsystemen für kurze Zeit und in geringem Umfang möglichst nur eine Cyclooxygenase blockiert wird. Im Umkehrschluss bedeutet dies, zur Schmerztherapie sollten möglichst lokal begrenzt, kurzfristig und selektiv wirkende Cyclooxygenasehemmer zum Einsatz kommen.
Es ist wichtig festzuhalten, dass alle genannten Wirkstoffe in analgetischer Dosierung beide Cyclooxygenasen hemmen. Diclofenac und Paracetamol zeichnen sich durch eine gewisse Selektivität für die COX-2 aus [29, 16], während ASS – und dieses ist eine besondere Eigenschaft dieses Wirkstoffes – die in den Blutplättchen lokalisierte COX-1 durch kovalente Bindung für die gesamte Lebensdauer des Blutplättchens (ca. 10 Tage) außer Gefecht setzt [10]. Daraus resultiert die antithrombotische Wirksamkeit der ASS, denn Blutplättchen können nicht, wie z. B. Endothelzellen, neue Cyclooxygenasen bilden und damit innerhalb von Stunden die irreversible Blockade der COX-1 kompensieren. Auch Naproxen kann die COX-1 so nachhaltig blockieren, dass eine messbare Einschränkung der Blutgerinnung auftritt, allerdings in geringerem Umfang als bei ASS [5, 14]. Dass die präferentielle Hemmung der COX-2 durch Paracetamol und Diclofenac zur Verträglichkeit dieser Wirkstoffe (Magenverträglichkeit, keine Blutgerinnungsstörungen) beiträgt, darf vermutet werden, ist aber nicht abschließend geklärt.
Wirkorte
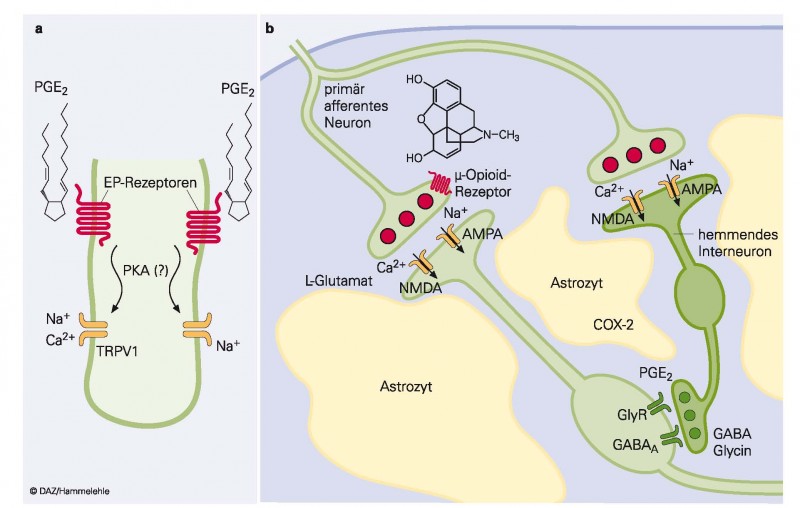
Hauptwirkort aller OTC-Analgetika ist zum einen das entzündete, traumatisierte oder infizierte Gewebe, von dem ein akuter Schmerzreiz ausgeht (Abb. 1a). Dort wird die Empfindlichkeit der Nozizeptoren durch Prostaglandine erhöht, sodass sie bereits bei normalerweise nicht schmerzhaften Reizen, wie Wärme bis 40 °C oder leichter Druck, mit einer schmerzhaften Wahrnehmung reagieren [2]. Es liegt auf der Hand, dass die Hemmung der Prostaglandine im entzündeten, traumatisierten oder chirurgisch veränderten Gewebe zu einer Normalisierung der Empfindlichkeit der Nozizeptoren führt (Antihyperalgesie).
Abb. 1:Physiologie des Schmerzes. a) Nozizeptor mit zwei Signaltransduktionswegen, von denen vermutet wird, dass sie zu peripherer Schmerzsensibilisierung führen. Prostaglandin E2 führt über EP-Rezeptoren zur Phosphorylierung und erleichterten Aktivierung des Hitzerezeptors TRPV1 und Tetrodotoxin-resistenter Natriumkanäle. b) Synaptische Verschaltungen im Hinterhorn des Rückenmarks. Primär afferente, nozizeptive Neuronen gehen syn-aptische Kontakte mit zentralen Projektionsneuronen und erregenden und hemmenden Interneuronen ein. Die pri-mären Afferenzen und die erregenden Interneuronen setzen L-Glutamat als erregenden schnellen Transmitter frei, während die hemmenden Neuronen Glycin und GABA als Überträgerstoffe verwenden. Durch ein peripheres Trauma (Entzündung usw.) wird durch Induktion von COX-2 vermehrt PGE2 gebildet. PGE2 blockiert den Glycinrezeptor (GlyR) und verhindert so die körpereigene Schmerzunterdrückung. Eine COX-2-Blockade rekonstituiert die glycinerge Schmerzhemmung [24].
Quelle: BIOspektrum 2004;1:36 – 38
Einen weiteren Wirkort hat die Forschung im Hinterhorn des Rückenmarks identifiziert (Abb. 1b). Auch hier führt ein peripheres Trauma zur vermehrten Prostaglandinbildung. Dabei wird durch chemische Mediatoren (Zytokine) sowie neuronale Depolarisationen im Hinterhorn des Rückenmarks die Prostaglandinsynthese erhöht (ebenfalls unter Induktion des Enzyms COX-2 [26]). Dadurch wird die Inhibition der Übertragung vom ersten Schmerzneuron auf das zweite mithilfe glycinerger Interneuronen aufgehoben (Desinhibition), sodass Schmerzreize ungefiltert höhere kognitive Zentren erreichen und als Schmerz wahrgenommen werden [24]. Diese Mechanismen sind grundsätzlich gleich. Die Intensität des Effektes in der Peripherie und im Zentralnervensystem ist aber unterschiedlich für die verschiedenen Wirkstoffe. Entscheidend ist daher nicht nur ihr Wirkungsmechanismus, sondern auch ihr pharmakokinetisches Verhalten.
Die Wirkpotenz scheint bis auf eine Ausnahme (Paracetamol) unerheblich. Da Paracetamol nur ein sehr schwacher Hemmer der Prostaglandinproduktion ist, der noch dazu sehr schnell metabolisiert wird (t50% = 1–2 Stunden; vgl. Tab. 1), müssen Einzeldosen von 1 g oft mehrfach wiederholt gegeben werden, um einen messbaren und nachhaltigen Effekt zu erzielen. Die Tagesdosis darf aber nur bis auf 4 g gesteigert werden, weil sonst lebensbedrohliche Leberschäden auftreten. So reicht die erzielbare analgetische Wirkung oft nicht zur Schmerzlinderung aus. Dann wird häufig ein Wirkstoff mit stärkerer Hemmpotenz (Diclofenac, Ibuprofen) eingesetzt. Das Paradoxon besteht darin, dass Paracetamol als besonders harmlos gilt, weil jeder Arzt und Apotheker weiß, dass es besonders toxisch ist und deshalb auf keinen Fall hoch dosiert werden darf [3].
Tab. 1: Pharmakologische und toxikologische Kenndaten von Schmerzmitteln. t
max
= maximaler Plasmaspiegel nach Applikation; t
50%
= Eliminationshalbwertszeit; BV
oral
= Bioverfügbarkeit nach oraler Gabe; V
D
= Verteilungskoeffizient.
| |||||
Substanz |
tmax (h)1
|
t50% (h) |
BVoral (%) |
VD
(l/kg) |
Risiko/Besonderheiten |
Säuren |
|||||
|
Acetylsalicylsäure
Salicylsäure2
|
0,5 – 2
0,5 – 2
|
~ 0,25
~ 1 – 33
|
~ 10
~ 90
|
? 4
~ 0,15
|
Hemmung der Plättchenaggregation ~ 5 Tage
Überdosierung: Lebensgefahr (besonders Kinder)
|
Diclofenac |
0,5 – 2 |
1 – 2 |
~ 60 |
~ 0,15 |
bei OTC-Dosierung Ø
bei antirheumatischer Dosierung wie andere NSAR
|
Ibuprofen |
0,5 – 2 |
1 – 2 |
~ 90 |
~ 0,15 |
bei OTC-Dosierung Ø
bei antirheumatischer Dosierung wie andere NSAR
|
Naproxen |
0,5 – 2 |
12 – 15 |
~ 90 |
~ 0,15 |
erhöhte Blutungsneigung
Ulzerationen im unteren Darmbereich
|
Nicht-Säuren |
|||||
Paracetamol |
0,5 – 2 |
1 – 2 |
~ 70 |
1,0 |
hepatotoxisch; bei Überdosierung letal |
Phenazon |
0,5 – 2 |
10 – 20 |
~ 90 |
1,0 |
schlechte Datenlage, allergische Reaktionen häufig (?)
zahlreiche Arzneimittelinteraktionen
|
Propyphenazon |
0,5 – 2 |
1 – 3 |
~ 90 |
1,0 |
schlechte Datenlage, allergische Reaktionen häufig (?) |
1
je nach galenischer Formulierung, 2
Salicylsäure ist der Hauptmetabolit, 3
bei Dosen > 0,6 g: dosisabhängig | |||||
Toxikologische Besonderheiten
Die meisten unerwünschten Arzneimittelwirkungen der Cyclooxygenasehemmer resultieren aus dem (für die Schmerzlinderung erwünschten) Fehlen der Prostaglandine: Hemmung der Plättchenaggregation, Blockade der gastroduodenalen Protektion, Verminderung des Schutzes der Lungenschleimhaut bei chronischen, entzündlichen Veränderungen des Respirationstraktes, Hemmung des hyperosmolaren Schutzes der Nierentubuli – um nur die wichtigsten zu nennen. Diese Risiken treffen auf alle Wirkstoffe zu; so werden Nierenschäden beim langfristigen Gebrauch für alle Wirkstoffe postuliert. Allerdings ist das Auftreten zahlreicher gastrointestinaler Risiken bei OTC-Dosierung (zur Selbstbehandlung von Schmerzen) nur für ASS und Naproxen gesichert [19]; asthmaähnliche, (pseudo-) allergische Reaktionen sind bei Paracetamol seltener als bei anderen, nicht-selektiven COX-Hemmern [28].
Darüber hinaus gibt es auch einige substanztypische, nicht cyclooxygenaseabhängige Toxizitätsprobleme. Sie betreffen vor allem die Leber. Hier kommt es besonders unter Paracetamol häufig [20], bei Acetylsalicylsäure nach Überdosierung und bei Diclofenac nach langfristiger Einnahme antirheumatischer Dosen [1, 25] gelegentlich zu Leberschäden; bei OTC-Dosierung von Diclofenac sind Leberschäden bisher nicht aufgetreten.
Das Risiko einer toxischen, ja sogar letalen Überdosierung von Acetylsalicylsäure bei Kindern ist geringer, seitdem dieser Wirkstoff kindersicherer verpackt und verwahrt wird. Durch Ibuprofen und Diclofenac allein sind auch bei massiver Überdosierung keine Todesfälle gemeldet worden [13]. Mit Paracetamol kann es zu lebensbedrohlichen oder sogar letalen Leberschäden kommen, da dieser Wirkstoff nur schwach analgetisch wirkt und vom Laien unter Umständen sowohl als Schmerzmittel als auch als Erkältungsmittel eingenommen wird und somit ungewollt überdosiert werden kann.
Pharmakokinetische Eigenschaften
Die analgetischen Wirkstoffe können aufgrund ihrer physikochemischen Eigenschaften grundsätzlich in zwei verschiedene Gruppen unterteilt werden:
- saure Wirkstoffe (ASS, Diclofenac, Ibuprofen und Naproxen) und
- nicht-saure Wirkstoffe (Paracetamol, Phenazon, Propyphenazon).
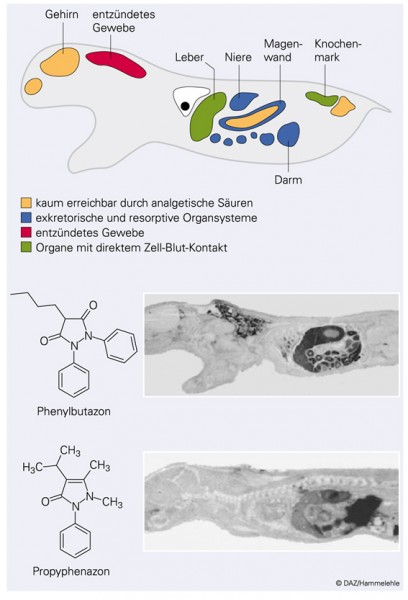
In beiden Gruppen gibt es Wirkstoffe mit sehr schneller und andere mit langsamer Elimination (Tab. 1). Grundsätzlich aber unterscheiden sich die beiden Gruppen darin, dass die nicht-sauren im menschlichen und tierischen Organismus homogen verteilt werden und die sauren besonders hohe Konzentrationen in bestimmten Körperregionen erreichen (Abb. 2). Mit anderen Worten: Die apolaren (nicht-sauren) Wirkstoffe weisen einen Verteilungskoeffizienten (VD) von etwa 1 l/kg auf, während die anderen aufgrund ihrer hohen Konzentrationen in Blut (inkl. Gefäßwand), Herz, Niere und Leber sowie dem entzündeten Gewebe einen VD von ca. 0,15 l/kg zeigen (Tab. 1, Abb. 2).
Abb. 2: Autoradiographische Analyse der Verteilung chemisch verwandter, aber unterschiedlich „saurer“ Wirkstoffe, nämlich des Phenylbutazons (Säure, pKa-Wert 4,5) und des Propyphenazons (schwache Base, pKa-Wert ca. 5). Bei gleicher Cyclooxygenasehemmpotenz wirkt Phenylbutazon stärker antiphlogistisch als Propyphenazon, weil es im entzündeten Gewebe akkumuliert (starke Schwarzfärbung im Bild); es provoziert aber auch Nierenfunktionsstörungen, Magen-Darm-Ulzerationen und interferiert mit der Blutgerinnung. Propyphenazon verteilt sich gleichmäßig im Organismus; die starke Schwarzfärbung weist zwar auf hohe Arzneistoffkonzentrationen im Nierenbecken, der Blase und dem Enddarm hin; es handelt sich aber um inaktive Metaboliten des Propyphenazons. Entsprechend der Verteilung wirkt Propyphenazon nur analgetisch (und kaum antiphlogistisch); es zeigt praktisch keine gastro-intestinalen und keine klinisch relevanten renalen und kardiovaskulären Nebenwirkungen, diffundiert aber schnell ins Rückenmark und bedingt durch COX-2 Hemmung (Abb. 1b) Schmerzlinderung.
Quelle: modifiziet aus: Brune K. Persistence of NSAIDs at effect sites and rapid disappearance from side-effect compartments contributes to tolerability.Curr Med Res Opin 2007;23(12):2985-95.
Diese Unterteilung erhält ihre besondere Bedeutung dadurch, dass die gewebeprotektiven, aber auch pro-algetischen Prostaglandine grundsätzlich in allen Körperzellen mit Ausnahme der roten Blutkörperchen synthetisiert und überall konzentrationsabhängig von Cyclooxygenase-Hemmstoffen unterdrückt werden [10]. Mit den apolaren Cyclooxygenasehemmern ist eine suffiziente Hemmung der Prostaglandinsynthese am Wirkort nur dann erzielbar, wenn gleichzeitig in allen anderen Geweben ebenfalls eine vergleichbare Prostaglandinsynthesehemmung stattfindet. Die sauren Cyclooxygenasehemmer dagegen hemmen die Bildung der Prostaglandine dort, wo sie sich anreichern. Alle anderen Gewebe sind bei Anwendung dieser Wirkstoffe keiner wesentlichen Prostaglandinsynthesehemmung unterworfen. Nur aufgrund der ungleichen Verteilung ist es möglich, durch Diclofenac die Prostaglandinsynthese im entzündeten Gewebe fast komplett zu unterdrücken [22] und einen durch Paracetamol oder Phenazon nicht erzielbaren antiphlogistischen Effekt zu erzielen.
Anreicherung im entzündeten Gewebe
In der täglichen Praxis haben sich nicht-selektive, saure Cyclooxygenasehemmer als unterschiedlich gastrointestinal-toxisch erwiesen. Als besonders gefährlich gelten das Phenylbutazon und das Piroxicam – beides Wirkstoffe mit langsamer Elimination und intensiver enterohepatischer Zirkulation. Sie sind beide wegen ihrer relativ hohen Toxizität verschreibungspflichtig [12].
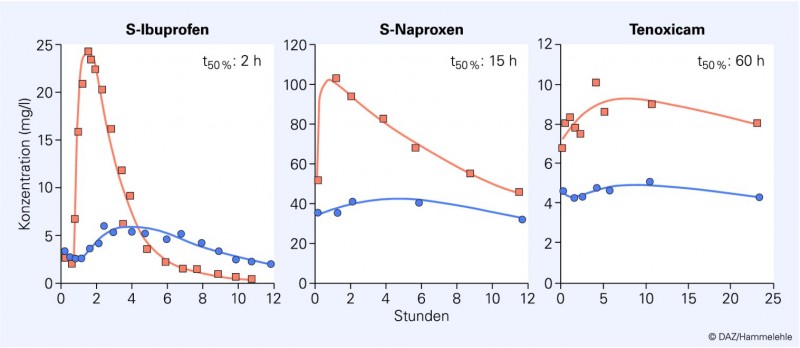
Die drei rezeptfrei verfügbaren Wirkstoffe Diclofenac, Ibuprofen und Naproxen akkumulieren alle im entzündeten Gewebe. Ein Vergleich der Pharmakokinetik von Ibuprofen, Naproxen und Tenoxicam bei Rheumapatienten zeigt, dass Ibuprofen im entzündeten Gelenk länger in hohen, wirksamen Konzentrationen präsent ist als im Plasma (Abb. 3) [7]. Dieses gilt nicht für Naproxen und schon gar nicht für Tenoxicam, die beide aufgrund ihrer erheblich längeren Halbwertszeiten sowohl im Plasma als auch im entzündeten Gewebe in hohen, COX-hemmenden Konzentrationen andauernd vorhanden sind. Diclofenac verhält sich wie Ibuprofen [9].
Abb. 3: Präsenz der Wirkstoffe Ibuprofen, Naproxen und Tenoxicam im Plasma (orange) und im entzündeten Gelenk (blau) von Rheumapatienten (nach [7]). Das kurzhalbwertszeitige Ibuprofen ist noch in wirksamer Konzentration im entzündeten Gewebe (Wirkort) vorhanden, wenn im Plasma schon unwirksame Konzentrationen erreicht werden. Es ist offensichtlich, dass bei zweimal täglicher Gabe von Ibuprofen in Dosen von 400 oder 600 mg eine Erholung im zentralen Kompartiment (Plasma) wahrscheinlich ist, während die Prostaglandinsynthesehemmung im entzündeten Gewebe fortbesteht. Eine persistierende Hemmung der Prostaglandinsynthese erfolgt natürlich auch nach der Gabe von 500 mg Naproxen oder 20 mg Tenoxicam. Allerdings kommt es aufgrund der langsamen Elimination aus dem Plasma nicht zu einer vergleichbaren Erholung der Prostaglandinsynthese. Es lässt sich ebenfalls interpolieren, dass bei der Gabe der Maximaldosis von Ibuprofen (2,4 g/Tag in der Rheumatherapie; das Gleiche gilt für Diclofenac 150 mg/Tag) keine Erholung der Prostaglandinsynthese in Herz, Niere, Plasma und Gefäßwand zu erwarten ist. Kardiovaskuläre Risiken werden dann manifest.
Die schnelle Elimination von Diclofenac und Ibuprofen führt zu einem interessanten pharmakokinetischen Phänomen, dem "crossing over" der beiden Konzentrationskurven (Abb. 3). Es erklärt
- die Wirksamkeit und Harmlosigkeit von Diclofenac und Ibuprofen in niedriger Dosierung sowie
- die im Verhältnis zur Eliminationshalbwertszeit lange Wirkdauer einer Einzeldosis.
Naproxen hat diese Vorteile nicht – aufgrund der deutlich langsameren Elimination. Piroxicam und Tenoxicam sind mit noch längeren t50% -Werten sicher noch weniger geeignet. In der Tat fallen diese Wirkstoffe bei großen epidemiologischen Untersuchungen negativ auf, während Diclofenac und Ibuprofen verhältnismäßig sicher sind [12].
Schließlich konnte gezeigt werden, dass bei zweimal täglicher Diclofenac-Applikation in der Entzündungsflüssigkeit eine permanente, fast totale Hemmung der Cyclooxygenasen erfolgt – trotz der kurzen Halbwertszeit des Wirkstoffes [22]. Allerdings wurden diese Untersuchungen mit relativ hohen, für die Selbstmedikation nicht zugelassenen Dosen durchgeführt. Sie können auch im Plasma zu einer anhaltenden, weitgehenden Hemmung der COX-2 führen [17].
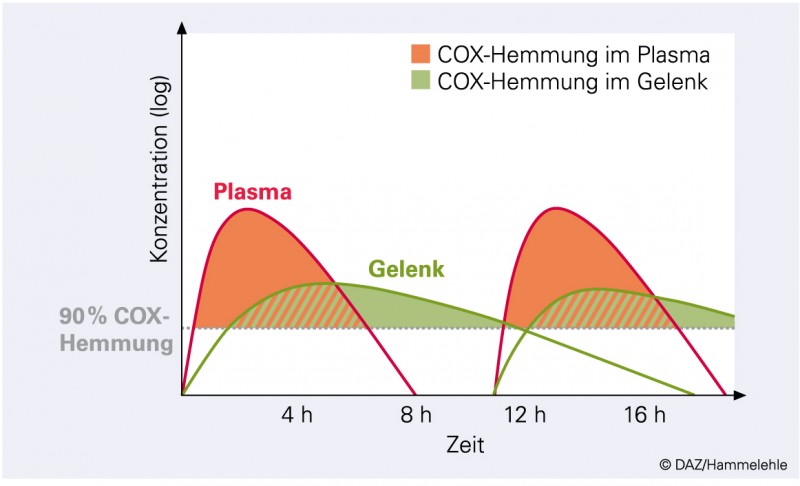
Es bleibt festzuhalten, dass sich die Synthese organoprotektiver Prostaglandine, z. B. des Prostacyclins der Gefäßwand oder des Prostaglandin E2 der Niere, bei zweimal täglicher Gabe von Diclofenac oder Ibuprofen in den für die Selbstmedikation zugelassenen niedrigen Dosierungen erholt, nicht jedoch bei der Gabe des langhalbwertszeitigen Naproxens [5, 14]. Bei Ibuprofen oder Diclofenac wird die Prostaglandinsynthese im zentralen Kompartiment unterbrochen, während sie im entzündeten Gewebe, dem Zielort, auf Dauer gehemmt bleibt (Abb. 4).*
| * Eine derartige Analyse der Wirkung von ASS ist nicht möglich, weil sie nach der Applikation nur kurzzeitig (Minuten) im Plasma nachweisbar ist, während im entzündeten Gewebe ihr Metabolit Salicylsäure auftaucht [23], deren antiphlogistisch-analgetische Wirksamkeit unklar bleibt. |
Abb. 4:
Idealisiertes Konzentrations-Zeit-Profil von kurzhalbwertszeitigen sauren Cyclooxygenasehemmern (vgl. Ibuprofen in Abb. 3). Bei zweimal täglicher Gabe wird eine nahezu permanente Hemmung der Prostaglandinsynthese im entzündeten Gewebe erreicht, während es im zentralen Kompartiment (Plasma) Erholungsphasen gibt.
Natürlich kommt es auch bei Gabe von 1 g Paracetamol zu einer Erholung der Prostaglandinsynthese im Gefäßsystem, gleichzeitig aber ebenfalls zu einem Abklingen der Prostaglandinsynthesehemmung am Wirkort. Bereits bei 4 × 1 g/Tag werden die Erholungsphasen nur sehr kurz sein. Dieses könnte erklären, warum bei langfristiger, hoch dosierter (4 g/Tag) Anwendung von Paracetamol ein erhöhtes kardiovaskuläres Risiko sichtbar wird [6]. Da weder Paracetamol noch Phenazon noch Propyphenazon im entzündeten Gewebe angereichert und retiniert werden, muss der Patient dauernd nachdosieren, um den analgetischen Effekt zu erhalten. Selbst dann wird die Prostaglandinsynthese im entzündeten Gewebe bei atoxischer Dosierung nur geringgradig unterdrückt. So findet auch die bekannte Beobachtung, nicht-saure Cyclooxygenasehemmer seien nicht antiphlogistisch wirksam, eine plausible Erklärung – wahrscheinlich beeinflussen sie nur die Prostaglandinsynthese im Rückenmark (vgl. Abb. 1b) in analgetisch ausreichendem Umfang.
Abschließende Bewertung
Bewertet man vergleichend die pharmakokinetischen, mechanistischen und toxikologischen Eigenschaften der rezeptfrei erhältlichen, analgetischen Wirkstoffe, so muss man feststellen, dass es keinen Wirkstoff gibt, der nicht auch Probleme aufwerfen kann. Allerdings müssen zwei Wirkstoffe als potenziell relativ toxisch bewertet werden, nämlich ASS und Paracetamol: Sie können beide bei Überdosierung zum Tode führen, und auch in der für die Selbstmedikation zugelassenen Dosierung lösen sie regelmäßig (ASS) bzw. häufig (Paracetamol) unerwünschte Wirkungen aus:
- ASS erhöht unvermeidlich das Blutungsrisiko,
- Paracetamol führt oft zu erhöhten Transaminasenwerten (ein Hinweis auf Leberschäden).
Beide Wirkstoffe behalten ihre Risiken auch in Kombinationspräparaten.
Paracetamol
Manche Fachleute halten Risiken aufgrund einer Überdosierung nicht für entscheidungsrelevant. (Schließlich kann man sich auch mit Insektiziden, Erdölprodukten oder Mineralien umbringen.) Hier ist aber festzustellen, dass Paracetamol bereits in therapeutischer Dosierung zu Leberschäden führen kann; solche Fälle sind wahrscheinlich viel häufiger als bisher bekannt. Immerhin weist bei viermal täglicher Gabe von 1 g fast ein Drittel der Probanden deutlich erhöhte Transaminasenwerte auf [30]. In Amerika gilt Paracetamol als die häufigste Ursache für Leberversagen [20]. Paracetamol ist also ein nur geringgradig wirksamer, letztlich nicht so harmloser Wirkstoff, wie der Laie oft glaubt. Er könnte fast immer durch Diclofenac oder Ibuprofen ersetzt werden. Als Ausnahme-Indikationen für niedrig dosiertes Paracetamol könnten Schwangere, Patienten mit pseudoallergischem Asthma oder floriden Magen-Darm-Ulzera gelten. Allerdings sind diese Patienten keine Zielgruppe der Selbstmedikation. Die Verkleinerung der Packungsgrößen auf 10 g in einigen Ländern der EU ist ein sinnvoller, aber nicht ausreichender Weg, die Risiken zu senken.
ASS
Anders liegt die Situation bei ASS. Schwere Blutungen, z. B. nach Zahnextraktionen, kommen immer wieder vor, weil dem Patienten nicht klar war, dass er zuvor niedrig dosierte ASS, z. B. in einem Kombinationspräparat, eingenommen hat. Nach Unfällen kann gelegentlich nicht oder nur unter Schwierigkeiten operiert werden, weil das Unfallopfer ASS zu sich genommen hatte und damit über keine ausreichende Blutgerinnung verfügte. ASS führt eindeutig auch in OTC-Dosierung zu Magen-Darm-Blutungen und Ulzerationen (evtl. auf der Basis der Gerinnungshemmung [11]). Da die Hemmung der Blutgerinnung durch ASS unvermeidlich (s. o.), zur analgetischen Wirkung aber nicht nötig ist (vgl. Diclofenac und Ibuprofen), sollte auf ASS in der Schmerz- genauso wie bereits jetzt in der Rheumatherapie verzichtet werden – zumal ASS bei Schwangeren (Blutungen), Kindern (Reye-Syndrom) und Patienten mit genuiner oder therapeutischer Blutgerinnungshemmung kontraindiziert ist. Die ASS hat ihren herausragenden Platz in der Infarktprophylaxe.
Phenazon
Phenazon muss als obsolet gelten. Bei gleichem Wirkmechanismus und ähnlichen Wissenslücken wie beim Propyphenazon sprechen die langsame Elimination sowie zahlreiche Arzneimittelinteraktionen gegen diesen Wirkstoff.
Naproxen
Auch Naproxen hat nicht im gleichen Umfang wie Diclofenac und Ibuprofen den Vorteil, im Dosisintervall eine Erholung der Cyclooxygenaseaktivität in Herz, Blut, Niere und Leber zu ermöglichen (Abb. 3 und 4). Diesen Wirkstoff können (gesunde) Patienten in der Selbstmedikation von akuten, typischerweise aber zeitlich sehr limitierten Schmerzzuständen (Dysmenorrhö [21] oder Migräneattacken) anwenden. Obwohl Naproxen grundsätzlich die Blutgerinnung durch nachhaltige Hemmung der COX-1 behindert, kommt es bei Dysmenorrhö doch zu einer Reduktion des Blutverlustes, vermutlich durch Blockade der prostaglandinabhängigen Gebärmutterkrämpfe [8].
Diclofenac und Ibuprofen
Für die gelegentliche Anwendung bei geringen bis mittelstarken Schmerzen im Zusammenhang mit Entzündungen, Verletzungen, Traumen, Verwundungen usw. scheint die Verwendung von Diclofenac und Ibuprofen z. B. zweimal täglich besonders sinnvoll. Auch hier ist allerdings zu bedenken, dass die Wirkstoffe nicht grundsätzlich ohne typische unerwünschte Arzneimittelwirkungen sind. Diclofenac kann bei höherer Dosierung zu Leberschäden führen, und Ibuprofen wird häufig in Zusammenhang mit allergischen Hautreaktionen gebracht. Bei höherer, nicht OTC-gemäßer Dosierung treten Magen-Darm-Störungen in den Vordergrund. So bleibt die Auswahl auch unter den beiden bestgeeigneten Wirkstoffen immer noch ein schwieriger Entscheidungsprozess, dem sich der Apotheker bei der Beratung widmen muss.
Literatur
[1] Boelsterli UA. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol 2003;192(3):307-22.
[2] Brune K, Zeilhofer HU. Antipyretic analgesics: basic aspects. In: McMahon SB, Koltzenburg M: Wall and Melzack’s Textbook of Pain, 2006, 6th edition. pp 459-469. Churchill Livingstone, Edinburgh.
[3] Brune K. Lebertoxizität von Paracetamol. Der Internist 2007;9:1036-1038.
[4] Brune K. The early history of non-opioid analgesics. Acute Pain 1997;1(1):33-40.
[5] Capone ML, et al. Human pharmacology of naproxen sodium. J Pharmacol Exp Ther 2007;322(2):453-60.
[6] Chan AT, et al. Nonsteroidal antiinflammatory drugs, acetaminophen, and the risk of cardiovascular events. Circulation 2006;113(12):1578-87.
[7] Day RO, et al. Pharmacokinetics of nonsteroidal anti-inflammatory drugs in synovial fluid. Clin Pharmacokinet 1999;36(3):191-210.
[8] Edgren RA, Morton CJ. Naproxen sodium for OB/GYN use, with special reference to pain states: a review. Int J Fertil 1986;31(2):135-42.
[9] Elmquist WF, Chan KK, Sawchuk RJ. Transsynovial drug distribution: synovial mean transit time of diclofenac and other nonsteroidal antiinflammatory drugs. Pharm Res 1994;11(12):1689-97.
[10] Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Discov 2003;2(3):179-91.
[11] García Rodríguez LA, Hernández-Díaz S, de Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiologic studies. Br J Clin Pharmacol 2001;52(5):563-71.
[12] Henry D, et al. Variability in risk of gastrointestinal complications with individual non-steroidal anti-inflammatory drugs: results of a collaborative meta-analysis. Br Med J 1996;312:1563–6.
[13] Hersh EV, Moore PA, Ross GL. Over-the-counter analgesics and antipyretics: a critical assessment. Clin Ther 2000;22(5):500-48.
[14] Hinz B, et al. Impact of naproxen sodium at over-the-counter doses on cyclooxygenase isoforms in human volunteers. Int J Clin Pharmacol Ther 2008;46 (in print).
[15] Hinz B, et al. Dipyrone elicits substantial inhibition of peripheral cyclooxygenases in humans: new insights into the pharmacology of an old analgesic. FASEB J 2007;21(10): 2343-51.
[16] Hinz B, Cheremina O, Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J 2008;22(2):383-90.
[17] Hinz B, Dormann H, Brune K. More pronounced inhibition of cyclooxygenase 2, increase in blood pressure, and reduction of heart rate by treatment with diclofenac compared with celecoxib and rofecoxib. Arthritis Rheum 2006;54(1):282-91.
[18] Ishikawa TO, et al. Imaging cyclooxygenase-2 (Cox-2) gene expression in living animals with a luciferase knock-in reporter gene. Mol Imaging Biol 2006;8(3):171-87.
[19] Kaufman DW, et al. Nonsteroidal anti-inflammatory drug use in relation to major upper gastrointestinal bleeding. Clin Pharmacol Ther 1993;53(4):485-94.
[20] Lee WM. Acetaminophen toxicity: changing perceptions on a social/medical issue. Hepatology 2007;46(4):966-70.
[21] Lethaby A, et al. Nonsteroidal anti-inflammatory drugs for heavy menstrual bleeding. Cochrane Database Syst Rev 2007;(4):CD000400.
[22] Liauw HL, et al. Effects of Voltaren on arachidonic acid metabolism in arthritis patients. Agents Actions Suppl 1985;17:195-9.
[23] Rainsford KD, Schweitzer A, Brune K. Distribution of the acetyl compared with the salicyl moiety of acetylsalicylic acid. Acetylation of macromolecules in organs wherein side-effects are manifest. Biochem Pharmacol 1983;32(7):1301-8.
[24] Reinold H, et al. Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. J Clin Invest 2005;115(3):673-9.
[25] Rubenstein JH, Laine L. Systematic review: the hepatotoxicity of non-steroidal anti-inflammatory drugs. Aliment Pharmacol Ther 2004;20(4):373-80.
[26] Samad TA, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001;410(6827):471-5.
[27] Simmons DL, Botting RM, Hla T. Cyclooxygenase isoenzymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 2004;56(3):387-437.
[28] Szczeklik A, et al. Analgesics and asthma. Am J Ther 2002;9(3):233-43.
[29] Tegeder I, et al. Comparison of inhibitory effects of meloxicam and diclofenac on human thromboxane biosynthesis after single doses and at steady state. Clin Pharmacol Ther 1999;65(5):533-44.
[30] Watkins PB, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006;296(1):87-93.
Anschrift für die Verfasser:
Prof. Dr. med. Dr. h. c. Kay Brune
Institut für Experimentelle und Klinische Pharmakologie und Toxikologie
Universität Erlangen-Nürnberg
Fahrstr. 17, 91054 Erlangen
Literaturtipp
Der "Zenz/Jurna" hält die Antworten auf die häufigsten Fragen in der Schmerztherapie bereit. Zum Beispiel:
- Welcher Schmerz macht welche Symptome?
- Wie wirken Analgetika?
- Wann wird Schmerz chronisch?
Zenz, Michael (Hrsg.) / Jurna, Ilmar (Hrsg.):
Lehrbuch der SchmerztherapieGrundlagen, Theorie und Praxis für Aus- und Weiterbildung
2. Auflage. XIV, 970 Seiten, 324 s/w Abb., 249 Tab. Geb. 75,70 Euro
Wissenschaftliche Verlagsgesellschaft, Stuttgart 2001, ISBN 978-3-8047-1805-0
Dieses Buch können Sie einfach und schnell bestellen unter der Postadresse:
Deutscher Apotheker Verlag, Postfach 10 10 61, 70009 Stuttgart
oder im Internet unter: www.dav-buchhandlung.de
oder per Telefon unter: (07 11) 25 82 - 3 41
oder- 3 42



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.