


















- DAZ.online
- DAZ / AZ
- DAZ 53/2020
- Informationen überunerw...
Arzneimittelsicherheit
Informationen überunerwünschte Wirkungen
Risiko einer Herzklappeninsuffizienz unter Fluorchinolonen
Mit einem Rote-Hand-Brief wurde über das Risiko einer Herzklappeninsuffizienz unter systemischer und inhalativer Anwendung von Fluorchinolon-haltigen Arzneimittel informiert. In Deutschland betrifft das Ciprofloxacin, Levofloxacin, Moxifloxacin, Norfloxacin, Ofloxacin und Delafloxacin. Die Breitbandantibiotika werden gegen bestimmte, auch lebensbedrohliche, bakterielle Infektionen eingesetzt. In einer epidemiologischen Studie wurde bei Patienten mit systemischer Fluorchinolon-Therapie ein etwa zweifach erhöhtes Risiko für eine Mitral- und Aortenklappeninsuffizienz im Vergleich zu Patienten beobachtet, die andere Antibiotika (Amoxicillin oder Azithromycin) einnahmen. Es gibt Hinweise, dass es einen Zusammenhang mit einem Fluorchinolon-assoziierten Abbau des Bindegewebes geben könnte. Fluorchinolone sollten nur nach sorgfältiger Nutzen-Risiko-Abwägung bei Patienten mit einem Risiko für eine Herzklappeninsuffizienz eingesetzt werden. (DAZ 44, S. 105)
Foto: Superingo – stock.adobe.com
Nur unter ärztlicher Aufsicht In die Produktinformationen zu Otriven® 0,025% Nasentropfen wurde der Hinweis aufgenommen, dass die Anwendung bei Kindern unter einem Jahr nur unter ärztlicher Aufsicht erfolgen sollte.
Kein Otriven®gegen Schnupfen bei Kleinkindern unter einem Jahr
Um eine mögliche Fehlanwendung und eine damit verbundene Überdosierung bei Säuglingen unter einem Jahr zu vermeiden, beschränkt die GlaxoSmithKline Consumer Healthcare GmbH & Co. KG die Anwendung von Xylometazolin 0,025% Nasentropfen (Otriven®gegen Schnupfen) auf Kleinkinder im Alter zwischen einem und zwei Jahren. Hintergrund ist, dass der AMK potenzielle Medikationsfehler berichtet wurden, wonach die Pipette zum Applizieren der Tropfen unter Umständen zu Fehldosierungen beitragen kann. Eine zuverlässige Gabe der empfohlenen Menge an Nasentropfen sei mit der beiliegenden Pipettenmontur schwierig umzusetzen, insbesondere bei der Applikation an unruhige Kinder. Im November 2020 wurde Ware mit der neuen Altersangabe in Deutschland eingeführt. Die Firma plant, nach Entwicklung eines neuen Applikators die Anwendung bei unter Einjährigen wieder zu ermöglichen. Die Produktinformationen abschwellender Xylometazolin- und Oxymetazolin-haltiger Nasentropfen zur Anwendung bei Säuglingen und Kleinkindern enthalten einen Warnhinweis, der über schwere Nebenwirkungen wie Atemstillstand bei Neugeborenen und jungen Säuglingen informiert. Diese können bei dieser Patientengruppe bereits bei der Applikation von therapeutischen Dosen, zwei- bis dreimal täglich ein Tropfen in jede Nasenöffnung, auftreten. (DAZ 4, S. 93, DAZ 46, S. 96)
Foto: Loocid GmbH – stock.adobe.com
Vor der Erstverordnung von Fingolimod sollte der Impfstatus des Patienten erfragt werden, jegliche Anzeichen einer möglicherweise schweren Infektion sollten unverzüglich dem behandelnden Arzt mitgeteilt werden.
Risiko für eine Meningoenzephalitis im Zusammenhang mit Fingolimod
Die Arzneimittelkommission der Deutschen Ärzteschaft (AkdÄ) berichtete über eine Patientin, die seit über 20 Jahren an schubförmiger multipler Sklerose (MS) leidet und eine Varizella-Zoster-Virus(VZV)-Meningoenzephalitis entwickelte. Seit fünf Jahren war die Patientin auf Fingolimod (Gilenya®) eingestellt. Der Allgemeinzustand der Patientin verschlechterte sich, es trat eine Sensibilitätsstörung im rechten Bein auf. Mit Verdacht auf einen MS-Schub wurde eine hochdosierte Stoß-Therapie mit intravenösen Corticosteroiden eingeleitet, ohne dass eine Besserung eintrat. Nach weiterer Diagnostik wurden meningoenzephalitische Veränderungen im MRT sowie Varizella-Zoster-Viren im Liquor nachgewiesen. Die AkdÄ nimmt diesen Fall zum Anlass, an das Risiko von VZV-Infektionen aufgrund der immunsuppressiven Wirkung von Fingolimod zu erinnern. Bereits in den Zulassungsstudien wurden Herpesvirus-Infektionen bei Patienten beschrieben. In den Produktinformationen wird von Patienten mit multipler Sklerose vor Therapiebeginn die Testung auf Immunität gegen Varizellen (Windpocken) und humanen Papillomaviren (HPV) gefordert. Sind diese nicht gegeben, ist eine vollständige Immunisierung vor der ersten Einnahme durchzuführen. (DAZ 48, S. 115)
Andexanet alfa nicht vor einer Heparinisierung anwenden
Das Antidot Andexanet alfa ist indiziert bei Erwachsenen, die mit einem direkten FXa-Inhibitor (Apixaban oder Rivaroxaban) behandelt werden, wenn aufgrund lebensbedrohlicher oder nicht kontrollierbarer Blutungen die Antikoagulation beendet werden muss. Werden Andexanet alfa und Heparin in kurzem Abstand zueinander gegeben, können die Ergebnisse von Gerinnungstests verfälscht werden. Andexanet alfa sollte vor einer Heparinisierung z. B. während eines chirurgischen Eingriffs vermieden werden, da dies zu einem Nicht-Ansprechen auf die gerinnungshemmende Wirkung von Heparin führen kann. Die Überwachung der Wirkung von Andexanet alfa in Gegenwart von aktivem Heparin wurde nicht validiert; auch die Anwendung zur Anti-FXa-Aufhebung vor einem dringenden chirurgischen Eingriff wurde nicht untersucht. (DAZ 26, S. 108, DAZ 46, S. 95)
Zusätzliche Charge Influenza-Impfstoff Vaxigrip Tetra aus Frankreich
Das PEI informierte über den Import zusätzlicher Packungen von Vaxigrip Tetra 2020/2021 aus Frankreich unter der Regelung der „Medizinischer Bedarf Versorgungssicherstellungsverordnung (MedBVSV)“. Die Ware ist in französischer Aufmachung und mit ausschließlich französischsprachigen Produktinformationen versehen. Die Packungen tragen einen Aufkleber mit eigener PZN und sind nicht serialisiert. Klebeetiketten zur Dokumentation der durchgeführten Impfung sind enthalten. Die deutschsprachigen Produktinformationen stehen in der ABDA-Datenbank unter dem Eintrag des deutschen Originalpräparates zur Verfügung. (DAZ 45, S. 113)
Kein Ulipristal zur Behandlung von Uterusmyomen
Im Rahmen des laufenden Risikobewertungsverfahrens zu Ulipristalacetat 5 mg, Tabletten (Esmya®) zur Behandlung von Uterusmyomen hat der CHMP empfohlen, die entsprechenden Zulassungen ruhen zu lassen. Während der Sicherheitsüberprüfung sollen auch keine neuen Patientinnen mehr mit betroffenen Arzneimitteln behandelt werden. Hintergrund der 2017 gestarteten Risikobewertung sind Berichte über das Risiko schwerer Leberschäden. Von den mehr als 900.000 Patientinnen, die seit der Zulassung im Jahr 2012 mit dem Progesteron-Rezeptor-Modulator aufgrund von Myomen behandelt wurden, wurden Fälle von schweren Leberschäden gemeldet, von denen fünf zu einer Lebertransplantation geführt haben. Angehörige der Heilberufe werden aufgefordert, Patientinnen, die noch mit Ulipristalacetat wegen Gebärmuttermyomen behandelt werden, über das Risiko zu informieren, um in Rücksprache mit dem Arzt die Behandlung zu beenden. Weiterhin sollen bis zu vier Wochen nach Ende der Therapie Leberfunktionstests durchgeführt werden. Gleichzeitig ist auf Anzeichen von Leberfunktionsstörungen (Übelkeit, Schmerzen unter dem rechten Rippenbogen, Anorexie und Gelbsucht) zu achten. Präparate zur Notfall-Kontrazeption mit 30 mg Ulipristalacetat (ellaOne® und andere) sind nicht betroffen, es gibt keine Hinweise auf eine Leberschädigung bei diesen Arzneimitteln. (DAZ 12, S. 114)
Rote-Hand-Brief zu Dimethylfumarat
Dimethylfumarat (magensaftresistente Hartkapseln, Tecfidera®) ist zur Behandlung von Erwachsenen mit schubförmig remittierender multipler Sklerose indiziert. Es wurden Fälle von progressiver multifokaler Leukenzephalopathie (PML) bei bestehender leichter Lymphopenie gemeldet. Zuvor war PML nur im Zusammenhang mit einer mäßigen bis schweren Lymphopenie bestätigt worden. Dimethylfumarat ist bei vermuteter oder bestätigter PML kontraindiziert und darf bei Patienten mit schwerer Lymphopenie nicht eingeleitet werden. Patienten sollten Betreuungspersonen über die Behandlung mit Tecfidera® informieren, da diese mögliche Symptome einer PML wie z. B. kognitive Beeinträchtigungen wahrnehmen könnten, die vom Patienten selbst nicht bemerkt werden. (DAZ 46, S. 95)
Fehlerhafte Dosierung bei Eylea®-Fertigspritzen
In der Schweiz wies die Bayer (Schweiz) AG auf die korrekte Vorbereitung und Injektion von Aflibercept Injektionslösung in einer Fertigspritze (Eylea®) hin. Jede Spritze enthält 177 μl Lösung, die jedoch nicht vollständig genutzt werden darf. In einem Vorbereitungsschritt werden überschüssige Lösung sowie Luftblasen verworfen. Dadurch wird eine korrekte Einzeldosis von 50 μl, entsprechend 2 mg Aflibercept, gewährleistet. Aufgrund von Meldungen zu kurzzeitigen, vorübergehenden Erhöhungen des Augeninnendrucks und reversiblen Visuseinschränkungen unmittelbar nach intravitrealer Injektion des Ophthalmikums vermutet der Zulassungsinhaber, dass ein zu großes Volumen appliziert wurde. Es wird an die in der Fachinformation detailliert beschriebene und illustrierte Anwendung der Eylea®-Fertigspritze erinnert. (DAZ 49, S. 126)
Leberschäden unter Fingolimod
Die Novartis Pharma GmbH informierte über risikominimierende Maßnahmen bei der Anwendung von Fingolimod Hartkapseln (Gilenya®), da arzneimittelinduzierte Leberschäden möglich sind. Der Sphingosin-1-Phosphat(S1P)-Rezeptormodulator ist zur Monotherapie der hochaktiven schubförmig-remittierend verlaufenden multiplen Sklerose zugelassen. Es wurden drei Berichte zu Lebertransplantationen aufgrund von Leberschäden bei Patienten berichtet, die mit Fingolimod behandelt wurden. Die Warnhinweise in der Fachinformation wurden entsprechend aktualisiert: Vor Therapiebeginn muss ein Leberfunktionstest durchgeführt werden, der in Monat 1, 3, 6, 9 und 12 nach Therapiebeginn und anschließend regelmäßig bis zwei Monate nach Therapieende zu wiederholen ist. Steigen die Lebertransaminasen ohne klinische Symptome, müssen die Leberwerte engmaschiger kontrolliert oder die Therapie unterbrochen werden. Gibt es Hinweise auf eine Leberschädigung (z. B. Müdigkeit, Appetitlosigkeit, Erbrechen, Beschwerden im rechten Oberbauch, dunkler Urin, gelbe Haut oder Skleren) sollten umgehend Leberfunktionstests durchgeführt werden. (DAZ 46, S. 94)
Foto: Rafael Ben-Ari – stock.adobe.com
Aus für Kava-Kava. Unzureichende Wirksamkeitsbelege für ethanolische Kava-Kava-Extrakte in der klinischen Anwendung bei Angststörungen und ein hepatotoxisches Risiko führten zu einer negativen Nutzen-Risiko-Bewertung.
Erneuter Widerruf der Zulassungen Kava-Kava-haltiger Arzneimittel
Ende Dezember 2019 hat das BfArM den Widerruf der Zulassungen Kava-Kava (Piper methysticum G. Forst., rhizoma)-haltiger sowie Kavain-haltiger Arzneimittel einschließlich homöopathischer Zubereitungen mit einer Endkonzentration bis einschließlich D4 angeordnet. Schon Ende 2001, nachdem zum Teil schwere Leberschäden durch Kava-Kava-Extrakte bekannt wurden, informierte das BfArM über die Absicht, die Zulassungen zu widerrufen, was 2002 mit sofortiger Wirkung erfolgte. Seitdem waren Zubereitungen aus Kava-Kava sowie Kavain in der Liste der bedenklichen Rezepturarzneimittel der AMK enthalten. 2015 war aufgrund eines Gerichtsurteils zwischenzeitlich die Zulassung von Kava-Kava-haltigen Arzneimitteln wieder gestattet, allerdings unter Sicherheitsauflagen. Der Ausschuss für pflanzliche Arzneimittel der EMA kam 2019 zu dem Schluss, dass keine Pflanzenmonographie der EU zur Verwendung von Piper methysticum G. Forst., rhizoma in Arzneimitteln erstellt werden kann. Grund sind die unzureichenden Wirksamkeitsbelege ethanolischer Kava-Kava-Extrakte für die Anwendung bei Angststörungen. Zusammen mit dem bekannten hepatotoxischen Risiko führte dies zu einer negativen Nutzen-Risiko-Bewertung. (DAZ 1/2, S. 87)
Leberschädigung unter Pirfenidon
Die Roche Pharma AG informierte mittels Rote-Hand-Brief über das Risiko schwerer Fälle von arzneimittelinduziertem Leberschaden unter Pirfenidon (Esbriet®), teilweise mit tödlichem Ausgang. Das Immunsuppressivum und Antifibrotikum ist indiziert zur Behandlung von leichter bis mittelschwerer idiopathischer pulmonaler Fibrose. Vor Beginn der Therapie müssen die hepatischen Transaminasen und der Bilirubin-Spiegel untersucht werden. Dies ist während der ersten sechs Monate der Behandlung jeweils monatlich und anschließend alle drei Monate zu wiederholen. Berichten Patienten über Symptome, die auf eine Leberschädigung hindeuten können (z. B. Müdigkeit, Appetitlosigkeit, Beschwerden im rechten Oberbauch, dunkler Urin oder Gelbsucht), sollten umgehend Leberfunktionstests durchgeführt werden. Wie in der Fachinformation angegeben, muss bei einem Anstieg der Lebertransaminasen die Dosis angepasst oder die Therapie abgesetzt werden. (DAZ 15, S. 101, DAZ 44, S. 101)
Ruhen der Zulassung von Ingenolmebutat
Die Europäische Kommission hat das vorläufige Ruhen der Zulassung von Ingenolmebutat Gel (Picato®) angeordnet, das Arzneimittel ist nicht mehr verkehrsfähig. Grund sind Bedenken über einen möglichen Zusammenhang zwischen der Anwendung und der Entwicklung von Hautkrebs. Patienten wird empfohlen, die Anwendung von Ingenolmebutat zur topischen Behandlung aktinischer Keratosen der Haut vorsichtshalber einzustellen. Angehörige der Heilberufe sollten Patienten dazu auffordern, auf Hautveränderungen zu achten und diese ärztlich abklären zu lassen. (DAZ 4, S. 94)
Foto: Africa Studio – Fotolia.com
Den Missbrauch von Cannabis-Arzneimitteln zu verhindern, dazu kann aufmerksames pharmazeutisches Personal beitragen.
Missbrauch Cannabis-haltiger Arzneimittel erkennen
Die Möglichkeiten zur Anwendung von Cannabis zu medizinischen Zwecken wurden erweitert. Dazu gehören Fertigarzneimittel wie Sativex® und Canemes® sowie Rezepturarzneimittel wie Dronabinol, Nabilon, Cannabis-Blüten und Cannabis-Extrakte. Die AMK betont, dass Apotheken eine besondere Verantwortung zukomme, damit Arzneimittelrisiken und eine missbräuchliche Anwendung von Cannabis vermieden werden können. Bislang wurden entsprechende Missbrauchsverdachtsfälle von Apotheken jedoch (noch) nicht an die AMK gemeldet. Daher möchte die AMK daran erinnern, dass das pharmazeutische Personal Hinweise auf einen kritischen Gebrauch bzw. Missbrauch geben könnte. Verdachtsmomente für Missbrauch können sein:
- Feststellung geänderter oder gefälschter Verordnungen
- Versuche, die Rezepturzubereitung zu beeinflussen, z. B. dass die Droge unverarbeitet abgegeben werden soll
- nicht medizinische Nutzung, z. B. eine zweifelhafte Gebrauchsanweisung
- Verordnung von mehreren (wohnortfernen) Ärzten
- Beschaffung aus mehreren (wohnortfernen) Apotheken.
Deuten die Patientenangaben auf einen Medikationsfehler, einen Missbrauch oder gar eine Abhängigkeit hin, so sollten individuell und abhängig vom jeweiligen Arzneimittel geeignete Lösungsmöglichkeiten angesprochen werden. Bei Ablehnung von Beratungsangeboten kann die Abgabe auch verweigert werden. (DAZ 3, S. 104)
Eingeschränkte Indikation bei Alemtuzumab
In einem Rote-Hand-Brief informierte die Firma Sanofi Belgium über Einschränkungen der Indikation, zusätzliche Gegenanzeigen und risikominimierende Maßnahmen bei Alemtuzumab Konzentrat zur Herstellung einer Infusionslösung (Lemtrada®). Aufgrund von Berichten über seltene, schwerwiegende Nebenwirkungen mit zum Teil tödlichem Ausgang war zuvor ein Risikobewertungsverfahren eingeleitet worden. Der monoklonale Antikörper ist angezeigt zur Monotherapie bei hochaktiver schubförmig-remittierender multipler Sklerose. Alemtuzumab soll u. a. nicht angewendet werden bei Patienten mit schweren aktiven Infektionen, unkontrollierter Hypertonie, Schlaganfall, Angina pectoris oder Myokardinfarkt, Koagulopathie, unter Therapie mit Thrombozytenaggregationshemmern oder Antikoagulanzien oder bei bestehenden Autoimmunerkrankungen, außer multiple Sklerose. Die Applikation sollte ausschließlich in Krankenhäusern mit der Möglichkeit einer sofortigen intensivmedizinischen Behandlung erfolgen. Für mindestens 48 Monate nach der letzten Infusion wird die Überwachung auf Autoimmunerkrankungen empfohlen. Patienten sollten darüber informiert werden, dass diese Erkrankungen auch nach mehr als 48 Monaten nach der letzten Infusion auftreten können. (DAZ 5, S. 96)
Risiko von Leberschäden unter Metamizol
In einem Rote-Hand-Brief wurde über Fälle eines arzneimittelbedingten Leberschadens unter Behandlung mit Metamizol berichtet. Das nichtopioide Pyrazolonderivat besitzt analgetische, antipyretische und spasmolytische Eigenschaften und wird bei akuten starken Schmerzen und hohem Fieber eingesetzt, wenn andere Maßnahmen nicht ansprechen. Anlässlich einer umfassenden Überprüfung durch den PRAC wurden Leberschäden identifiziert, die vorwiegend ein hepatozelluläres Muster zeigten und wenige Tage bis Monate nach Behandlungsbeginn auftraten. Bei einigen Patienten trat nach erneuter Anwendung wieder ein Leberschaden auf. Es wird ein immun-allergischer Pathomechanismus des Metamizol-bedingten Leberschadens vermutet. Er wird als sehr selten eingeschätzt, die genaue Häufigkeit kann nicht berechnet werden. Treten Symptome auf, die auf eine Leberschädigung hindeuten können, sollte die Anwendung von Metamizol beendet werden. (DAZ 51, S. 101)
Patientenkarte zum Risiko latenter BCG-Infektionen
Die Medac Gesellschaft für klinische Spezialpräparate mbH erinnert an das Risiko von Infektionen mit Bacillus Calmette-Guérin-Bakterien (BCG-Infektionen). BCG-medac zur Herstellung einer Suspension zur intravesikalen Anwendung enthält eine gefriergetrocknete Suspension lebender Bacillus Calmette-Guérin-Bakterien (BCG). Es wirkt als nichtspezifisches Immunstimulans und ist indiziert bei nichtinvasiven urothelialen Harnblasenkarzinomen. Die Therapie kann eine BCG-Infektion verursachen, die unter Umständen mehrere Jahre latent verläuft. Laut Einzelfallberichten kann es noch lange nach Ende der Therapie zu einem Aufflammen von latenten BCG-Infektionen kommen, die potenziell tödlich verlaufen könnten. Mit einer Patientenkarte, die u. a. die Symptome einer aufflammenden BCG-Infektion beschreibt, soll das Bewusstsein für dieses Risiko erhöht werden. Bis die Patientenkarte allen Packungen beiliegt, bittet die Firma, diese den Patienten mitzugeben. Bestellt werden können sie bei Medac Gesellschaft für klinische Spezialpräparate mbH, contact@medac.de. (DAZ 14, S. 109)
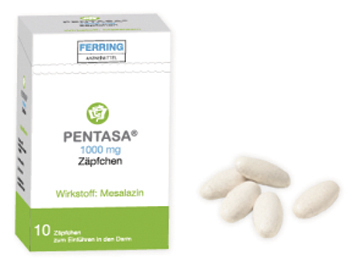
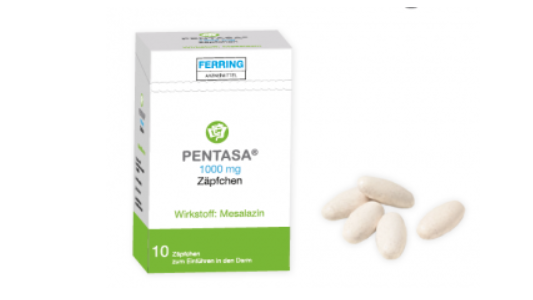
Foto: Ferring Arzneimittel
Pentasa® Zäpfchen sehen wie Tabletten aus. Die Mesalazin-haltigen Zäpfchen bestehen nicht aus Hartfett, sondern aus wasserunlöslichem Talkum und Magnesiumstearat. Das erfordert besondere Anwendungshinweise.
Risiko für Anwendungsfehler bei Mesalazin-haltigen Zäpfchen
Mesalazin-haltige Zäpfchen sind indiziert zur Behandlung einer Colitis ulcerosa, die sich auf den Enddarm beschränkt. Eine Analyse der AMK-Datenbank ergab insgesamt 30 Meldungen zu Pentasa® Zäpfchen. Die spezielle Galenik macht eine besondere Handhabung zum Einführen in das Rektum erforderlich. Daher kann es passieren, dass Zäpfchens im Analkanal anhaften und ein vermehrter Defäkationsreiz, lokales Brennen bis hin zu Mazerationen der Schleimhaut mit Blutbeimengungen auftreten. Häufig wurde berichtet, dass sich das Zäpfchen nicht „auflöse“. Die bei Pentasa® Zäpfchen verwendete Grundmasse besteht nicht aus Hartfett, sondern u. a. aus wasserunlöslichem Talkum und Magnesiumstearat. Das an eine Oblongtablette erinnernde Zäpfchen zerfällt daher nur im Rektum und bildet dort eine Suspension. Dieser Umstand kann zum Eindruck des „Nicht-Auflösens“ führen. In der Apotheke konnte bisher kaum zielgerichtet zur Anwendung beraten werden, weil weder die Bezeichnung des Arzneimittels oder die Umverpackung noch die Apothekensoftware – abgesehen von der hilfsstofflichen Zusammensetzung – hierzu Anlass boten. Erst bei der Entnahme des Zäpfchens aus dem Aluminium-Blister wurde erkennbar, dass es keine Hartfettzäpfchen sind, wie dies vom Produkt des Marktführers bekannt ist. Allerdings sind Apotheken seit der Änderung des Rahmenvertrags häufig verpflichtet, den Marktführer gegen Pentasa® Zäpfchen auszutauschen. Besonders Erstanwender von Pentasa® Zäpfchen (auch Importe) sollten diesbezüglich beraten werden. (DAZ 5, S. 96)
Anidulafungin Infusionslösung nicht einfrieren
Ein Rote-Hand-Brief informierte über eine geänderte Vorschrift zur Aufbewahrung von Anidulafungin Pulver zur Herstellung eines Konzentrats zur Herstellung einer Infusionslösung (Ecalta®). Entgegen der aktuellen Version der Produktinformationen (Stand September 2018) darf die Infusionslösung nicht eingefroren werden. Das Antimykotikum wird zur systemischen Behandlung invasiver Candidiasis bei Erwachsenen angewandt. Das Fertigarzneimittel muss vor der Anwendung mit Wasser für Injektionszwecke zu einem Konzentrat rekonstituiert werden. Anschließend wird das Konzentrat entweder mit isotonischer Kochsalzlösung oder 5%iger Glucoselösung zu einer Infusionslösung zur Anwendung am Patienten verdünnt. Stabilitätstests des Herstellers ergaben, dass nach Einfrieren und nachfolgendem Auftauen der Infusionslösung weiße, amorphe Wirkstoffpartikel auftreten können und die Lösung somit nicht verwendet werden darf. Die Infusionslösung darf wie bisher bei 25 °C für 48 Stunden aufbewahrt werden. Hergestellte Lösungen sind vor der Applikation optisch zu überprüfen. Werden Partikel und Verfärbungen festgestellt, ist die Lösung zu verwerfen. (DAZ 7, S. 87)
Systemische Nebenwirkungen bei vaginal appliziertem Linoladiol® N
Die Dr. August Wolff GmbH & Co. KG Arzneimittel informiert mit einem Rote-Hand-Brief zu Risiken systemischer Nebenwirkungen bei Applikation von Estradiol Creme zur vaginalen Anwendung (Linoladiol® N), die zur Behandlung vaginaler Atrophie aufgrund von Estrogen-Mangel bei postmenopausalen Frauen eingesetzt wird. Seit 2019 wird das Risiko der systemischen Aufnahme bei vaginaler Behandlung mit hochdosierten Zubereitungen mit 100 µg Estradiol pro g Creme untersucht. In pharmakokinetischen Studien zeigten sich nach intravaginaler Anwendung von Linoladiol® N Estradiol-Serumspiegel, die postmenopausale Serum-Referenzkonzentrationen von 10 bis 20 pg/ml Estradiol um das bis zu Fünffache überschreiten. Linoladiol® N sollte nicht gleichzeitig bei Patientinnen angewendet werden, die mit oralen oder transdermalen Arzneimitteln zur Hormonersatztherapie behandelt werden. (DAZ 9, S. 96)
Risiko für venöse thromboembolische Ereignisse unter Tofacitinib
Die Pfizer Pharma GmbH informiert mittels Rote-Hand-Brief zu einem dosisabhängig erhöhten Risiko für schwerwiegende venöse thromboembolische Ereignisse (Lungenembolien, tiefe Venenthrombosen) unter Tofacitinib (Xeljanz®). Das selektive Immunsuppressivum wird zur Behandlung der rheumatoiden Arthritis und Psoriasis-Arthritis sowie der Colitis ulcerosa eingesetzt. Da Patienten über 65 Jahre ein zusätzlich erhöhtes Risiko für schwerwiegende Infektionen sowie ein erhöhtes Mortalitätsrisiko aufgrund von Infektionen haben, wird der Einsatz bei Patienten mit Colitis ulcerosa und bekannten Risikofaktoren für thromboembolische Ereignisse nicht länger empfohlen, außer es steht keine geeignete Alternative zur Verfügung. Die empfohlene Dosis von zweimal täglich 5 mg sollte nicht überschritten werden. (DAZ 13, S. 99)
Wechselwirkung zwischen ASS und Metamizol
Anhand der periodischen Sicherheitsberichte der Zulassungsinhaber Acetylsalicylsäure(ASS)-haltiger Arzneimittel bewertete der PRAC unter anderem die Wechselwirkung zwischen ASS und Metamizol. Der PRAC war zur Auffassung gelangt, dass durch die gleichzeitige Einnahme von ASS und Metamizol in klinisch relevanter Weise die Hemmung der Thrombozytenaggregation durch ASS vermindert wird. Nun wurde der Wortlaut der Produktinformationen angepasst. Überdies sollten Patienten, die ASS in niedriger Dosierung zur Kardioprotektion einnehmen, Metamizol nur mit Vorsicht anwenden. (DAZ 14, S. 110)
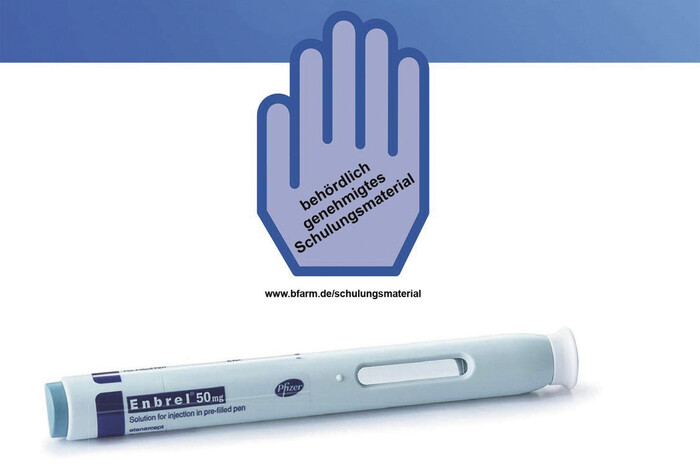
Foto: BfArM
Bei Verdacht auf einen Qualitätsmangel Etanercept-haltiger Pens oder Fertigspritzen sollte die korrekte Applikation durch den Patienten in der Apotheke erfragt werden. Schulungsmaterialien stehen auf der Homepage des BfArM zur Verfügung.
Fehlerhafte Injektionen bei Etanercept-haltigen Arzneimitteln
Etanercept (Enbrel®)ist ein biotechnologisch hergestelltes humanes Fusionsprotein, welches das proinflammatorische Zytokin Tumornekrosefaktor alpha (TNF-α) bindet und somit immunsuppressiv und entzündungshemmend wirkt und zur Behandlung der rheumatoiden Arthritis eingesetzt wird. Der TNF-α-Antagonist wird als Fertigpen, Fertigspritze oder als Trockensubstanz mit Lösungsmittel angeboten. Der AMK liegen seit Dezember 2012 bis Ende März 2020 insgesamt 217 Meldungen zu Etanercept-haltigen Arzneimitteln vor. Neben Meldungen zu unerwünschten Arzneimittelwirkungen stellen Berichte zu einem defekten Auslösemechanismus den Großteil der Meldungen dar. Davon wurden acht Fälle herstellerseitig als berechtigte Reklamation eingestuft. In den verbleibenden 124 war eine Überprüfung der Beanstandung entweder nicht möglich oder der Hersteller hat nach eigener Prüfung des eingesandten Musters keine Qualitätsmängel erkannt. In insgesamt 40 Fällen mit defektem Auslösemechanismus wurde ein Anwendungsfehler des Patienten vermutet. Um eine fehlerhafte Anwendung zu vermeiden, werden in den Gebrauchsinformationen konkrete Hinweise bebildert dargestellt, ebenso liegen zusätzlich Schulungsmaterialien (Blaue Hand) vor, die die korrekte Applikation des Arzneimittels beschreiben. (DAZ 30, S. 87)
Eingeschränkte Indikation für Cyproteronacetat
Die Zulassungsinhaber hochdosierter Cyproteronacetat-haltiger Arzneimittel informieren mittels Rote-Hand-Brief über das Risiko für Meningeome. Das antiandrogen wirkende Gestagen ist bei Frauen ab einer Dosis von 10 mg/Tag als Monotherapie bei mittelschweren bis schweren Androgenisierungserscheinungen indiziert. Bei Männern kann es u. a. zur palliativen Therapie des metastasierenden oder lokal fortgeschrittenen inoperablen Prostatakarzinoms eingesetzt werden. Aktuelle Ergebnisse einer epidemiologischen Kohortenstudie an rund 250.000 Frauen zeigten einen kumulativen, dosisabhängigen Zusammenhang zwischen der CPA-Einnahme und Meningeomen, die hauptsächlich bei Dosen von 25 mg Cyproteronacetat/Tag und darüber auftraten. Patienten unter CPA-Therapie sollten daher auf Meningeome überwacht werden. In der Therapie mit niedrig dosiertem CPA (1 bzw. 2 mg) in Kombination mit Estradiolvalerat bzw. Ethinylestradiol konnten keine neuen Sicherheitsbedenken identifiziert werden. Dennoch sind niedrig dosierte Kombinationsprodukte jetzt bei Patienten mit Meningeom oder mit Meningeom-Anamnese kontraindiziert. (DAZ 17, S. 115)
Äquivalenzdosistabellen als Hilfestellung zur Aut-simile-Auswahl
Mit Inkrafttreten der SARS-CoV-2-Arzneimittelversorgungsverordnung im April 2020 wurden Apotheken weitreichende Möglichkeiten eingeräumt, im Falle der Nichtverfügbarkeit eines verordneten Arzneimittels u. a. von der Packungsgröße, der Packungsanzahl oder der Wirkstärke abzuweichen. Ist kein wirkstoffgleiches Präparat verfügbar, kann nach Rücksprache mit dem Arzt ein pharmakologisch-therapeutisch vergleichbares Arzneimittel abgegeben werden (Aut-simile-Substitution). Dabei ist es in der Regel notwendig, zunächst die Dosisäquivalenz zum bisherigen Arzneimittel abzuschätzen. Als Hilfestellung veröffentlicht die AMK Vergleichstabellen zu Äquivalenz- bzw. Tagesdosen zu ausgesuchten Wirkstoffklassen. Diese stellen nur einen Anhaltspunkt dar, im Einzelfall sind Indikationen, Wechselwirkungen, Pharmakokinetik, Kontraindikationen sowie patientenindividuelle Faktoren zu berücksichtigen. Die Vergleichstabellen stehen auf der AMK-Homepage unter www.arzneimittelkommission.de bereit. (DAZ 18, S. 102)
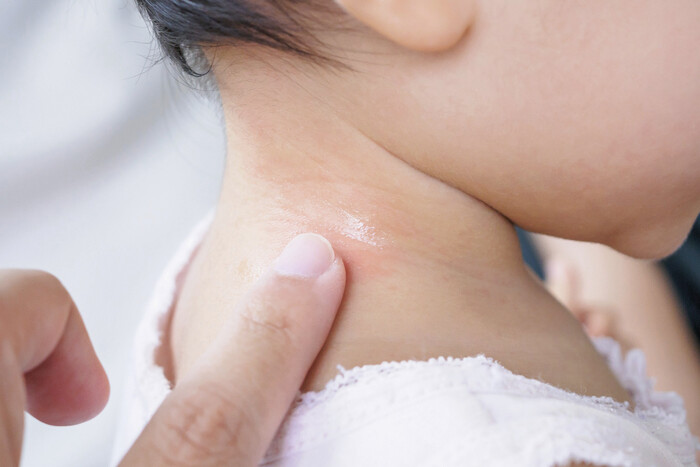
Foto: Piman Khrutmuang – stock.adobe.com
Äußerlich anzuwendende Lokalanästhetika können besonders bei Kindern gefährlich werden. Eltern sollten daher diese Produkte korrekt anwenden, um unerwünschte Arzneimittelwirkungen zu vermeiden.
Methämoglobinämie nach Überdosierung von Emla®-Creme
Die AkdÄ berichtet über einen Fall, in dem eine Methämoglobinämie nach einer Überdosierung von Emla® (Lidocain, Prilocain) 25 mg/g + 25 mg/g Creme bei einem sieben Monate alten Säugling festgestellt wurde, bei dem vier Wochen zuvor eine Zirkumzision durchgeführt worden war. Im Rahmen der postoperativen Wundpflege trugen die Eltern bei jedem Windelwechsel Emla®-Creme auf, die noch im Haushalt vorrätig war. Nach zwei Tagen entwickelte der Junge eine Zyanose. Ursächlich war eine Methämoglobinämie. Die Sauerstoffsättigung betrug minimal 73%. Auf der Intensivstation wurde Sauerstoff verabreicht, worunter die Sauerstoffsättigung stieg und sich stabilisierte. Die AMK erinnert auf das erhöhte Risiko von Nebenwirkungen, insbesondere bei Überdosierungen von äußerlich anzuwendenden, teilweise rezeptfreien Lokalanästhetika. Entsprechende Externa sollten nicht auf offenen Wunden oder potenziell geschädigter Haut angewendet werden, da es zu einer erhöhten systemischen Exposition kommen kann. Grundsätzlich besteht bei Personen mit einem Glucose-6-phosphat-Dehydrogenasemangel, vererbter oder idiopathischer Methämoglobinämie ein erhöhtes Risiko, eine arzneimittelinduzierte Methämoglobinämie zu entwickeln. Die AMK bittet Apotheker Anwender angemessen zu beraten. Insbesondere bei Anwendung an Kindern sollte darauf hingewiesen werden, die Empfehlungen zur Dosierung und zur Anwendungsdauer laut Produktinformation nicht zu überschreiten. (DAZ 21, S. 102)
Ruhen der Zulassungen Ranitidin-haltiger Arzneimittel
Der CHMP hat das Ruhen der Zulassungen aller Ranitidin-haltigen Arzneimittel in der EU empfohlen, da diese in geringen Mengen die potenziell krebserregende Verunreinigung N-Nitrosodimethylamin (NDMA) enthalten. In mehreren Ranitidin-haltigen Arzneimitteln liegen diese über den als akzeptabel angesehenen Werten. Die Quelle der Verunreinigung sei zum Teil noch unklar. Es gebe Hinweise darauf, dass sich NDMA aus dem Abbau von Ranitidin selbst mit über die (Lager-)Zeit zunehmenden Konzentrationen bilden kann. Auch sei noch ungewiss, ob NDMA endogen aus Ranitidin im Gastrointestinaltrakt gebildet werden könne. Verfügbare klinische und epidemiologische Daten zeigten kein erhöhtes Krebsrisiko bei Patienten, die mit Ranitidin behandelt wurden. Angesichts der vorhandenen Unsicherheiten hat der CHMP nun das vorsorgliche Ruhen der Zulassungen in der EU empfohlen. (DAZ 19, S. 96)
Rote-Hand-Brief zu Tolperison
Die Zulassungsinhaber Tolperison-haltiger Arzneimittel erinnern an die negative Nutzen-Risiko-Bewertung bei Off-Label-Anwendung. Ein 2013 vom BfArM initiiertes europäisches Risikobewertungsverfahren führte zur Indikationseinschränkung des zentral wirksamen Muskelrelaxans auf die symptomatische Behandlung von Spastizität bei Erwachsenen nach einem Schlaganfall. Für andere Anwendungsgebiete gibt es keinen ausreichenden Wirksamkeitsbeleg, zudem traten Überempfindlichkeitsreaktionen bis hin zu schweren anaphylaktischen Reaktionen auf. (DAZ 23, S. 99)
Tödliche Wechselwirkungen von Brivudin mit Fluoropyrimidinen
Ein Rote-Hand-Brief informierte über Maßnahmen, um Todesfälle aufgrund der bekannten Wechselwirkung zwischen Brivudin und Fluoropyrimidinen zu vermeiden. Das Nukleosid-Analogon Brivudin ist indiziert zur Behandlung des akuten Herpes Zoster bei immunkompetenten Erwachsenen. Als potenziell tödliche Wechselwirkung ist bekannt, dass das Virustatikum über seinen Hauptmetaboliten die Dihydropyrimidindehydrogenase (DPD) irreversibel hemmt und dadurch den Abbau von Fluoropyrimidinen verhindert. Es wird daran erinnert, dass Brivudin-haltige Arzneimitteln kurz vor, während und innerhalb von vier Wochen nach der Anwendung von Fluoropyrimidinen (das antineoplastisch wirksame Fluorouracil und seine Prodrugs Capecitabin und Tegafur sowie das Antimykotikum Flucytosin) kontraindiziert sind. Es wird zukünftig jeder Packung Brivudin-haltiger Arzneimittel eine neue Patientenkarte beiliegen, die Patienten bis mindestens vier Wochen nach dem Therapieende mit Brivudin bei jedem Arzttermin und Apothekenbesuch vorzeigen sollen. (DAZ 20, S. 100)
Foto: ia_64 – stock.adobe.com
Auch bei Dosieraerosolen stehen Devices mit Zählwerken zur Verfügung. Es kann jedoch bei Geräten mit Zählwerk durch verstopfte Düsen zu einer verschlechterten Symptomkontrolle kommen.
Verstopfte Düsen bei Dosieraerosolen mit Dosiszählwerk
Seitdem Foster®, Inuvair® und Trimbow® mit den Wirkstoffen Beclometason und Formoterol als Dosieraerosol mit Dosiszählwerk vermarktet werden, verzeichnet die AMK eine zunehmende Zahl an Spontanberichten aus Apotheken. Dosiszählwerke sollen durch die Anzeige der restlichen Dosen Patienten bei der korrekten Inhalation und Bevorratung unterstützen. Im Zeitraum von 2013 bis Ende 2019 wurden der AMK insgesamt 478 Verdachtsfälle über Risiken zu den Dosieraerosolen, einschließlich ihrer Importe (11%), gemeldet. Über die Hälfte der Meldungen (n = 277) bezog sich auf einen gestörten Auslösemechanismus. Insgesamt entfielen 64 Berichte auf Nebenwirkungsverdachtsfälle, wobei diese in 18 Fällen (13 allein in 2019) im engen zeitlichen Zusammenhang mit einer Fehlfunktion des Dosieraerosols auftraten. Zu vielen Fallberichten standen Reklamationsmuster für Untersuchungszwecke zur Verfügung. Die Eingangsprüfungen der AMK bestätigten vor allem weiße Ablagerungen an der Austrittsöffnung und am Ventilstift der Patrone sowie das Nichtauslösen der Dosieraerosole. Vermutet wird, dass die Löslichkeit des Beclometasondipropionats durch Verdunsten des Ethanols herabgesetzt wird und Wirkstoffkristalle an der engen Austrittsöffnung die Düse verschließen. Kristalle können sich auch innen an der Düse bilden. Das empfohlene Reinigungsverfahren in den Gebrauchsinformationen half in einigen Fällen nachweislich nicht, verschlossene Düsen wieder freizulegen. Die AMK gibt folgende Hinweise: Bei verminderter bzw. fehlender Aerosolabgabe ist der Inhalator mehrfach hintereinander auszulösen, bis wieder gleichmäßige Sprühstöße beobachtet werden können. Asthmatiker, die mit höheren Dosierungen bzw. der 200-µg-Beclometason-Stärke behandelt werden, sollten ab einem Zählerstand von etwa 50 Resthüben vor Beginn der Inhalation einen Sprühstoß in die Luft abgeben, um eine einwandfreie Aerosolbildung festzustellen. Asthmapatienten sollten bei Verwendung der Zweifachkombinationen ihren separaten schnellwirkenden Bronchodilatator für eine Notfallanwendung jederzeit griffbereit haben. Das Mundstück sollte zur Minimierung der Verdunstung des Ethanols mit der Kunststoffkappe verschlossen werden. Ferner erinnert die AMK daran, dass nach der ersten Anwendung die Verwendbarkeitsdauer auf drei Monate statt vormals fünf Monate verkürzt wurde. (DAZ 42, S. 107)
Sachverständigenausschuss für Verschreibungspflicht
Im Januar 2020 fand die 82. Sitzung des Sachverständigenausschusses für Verschreibungspflicht nach § 53 Absatz 2 des Arzneimittelgesetzes (AMG) statt. Unter anderem wurde folgenden Anträgen zur Änderung der Positionsformulierung zugestimmt:
„Lithium – zur Prophylaxe und/oder Behandlung von psychiatrischen Erkrankungen und Cluster-Kopfschmerzen –“.
Almotriptan und Naratriptan: „– ausgenommen zur akuten Behandlung der Kopfschmerzphase bei Migräneanfällen mit und ohne Aura, nach der Erstdiagnose einer Migräne durch einen Arzt, in festen Zubereitungen zur oralen Anwendung in Konzentrationen von 12,5 mg (bzw. 2,5 mg) je abgeteilter Form und in einer Gesamtmenge von 25 mg (bzw. 5 mg) je Packung.“ Die Altersbeschränkung wurde gestrichen. Almotriptan, Naratriptan und Sumatriptan 50 mg sind zur Anwendung bei Erwachsenen zwischen 18 und 65 Jahren vorgesehen.
Angenommen wurden die Anträge, Diphenhydramin und Doxylamin zur Behandlung von Schlafstörungen bei Patienten über 65 Jahren der Verschreibungspflicht zu unterstellen.
Abgelehnt wurde der Antrag auf Freistellung aus der Verschreibungspflicht für Eigenblut und Eigenblutzubereitungen humanen Ursprungs. (DAZ 5, S. 96)
Änderung in der Verschreibungspflicht
Im Februar wurde die 19. Verordnung zur Änderung der Arzneimittelverschreibungsverordnung (AMVV) im Bundesgesetzblatt veröffentlicht:
Die Position „Desloratadin“ wird neu gefasst: „– ausgenommen Arzneimittel in der oralen Anwendung zur symptomatischen Behandlung bei allergischer Rhinitis und Urtikaria bei Erwachsenen, Jugendlichen und Kindern ab zwei Jahren, es sei denn, es handelt sich um von der Europäischen Kommission als verschreibungspflichtig zugelassene Arzneimittel –“.
Wegen der unterschiedlichen Zulassungsarten (zentral/national) können sowohl verschreibungspflichtige als auch nicht verschreibungspflichtige Desloratadin-haltige Arzneimittel parallel im deutschen Markt verfügbar sein. (DAZ 10, S. 112)
Änderungen in der Verschreibungs- und Apothekenpflicht
Im Oktober 2020 wurde die Verordnung zur Änderung der Arzneimittelverschreibungsverordnung (AMVV), der Apothekenbetriebsordnung (ApBetrO) und der Verordnung über apothekenpflichtige und freiverkäufliche Arzneimittel (AMVerkRV) im Bundesgesetzblatt veröffentlicht. Die wichtigsten Änderungen sind u. a.:
- Auf einer Verordnung über intranasale Esketamin-haltige Arzneimittel muss vermerkt sein, dass die Abgabe nicht an den Patienten erfolgen darf. Apotheker dürfen einen fehlenden Vermerk nachtragen.
- „Sumatriptan“ wird für den Fall der „akuten Behandlung der Kopfschmerzphase bei Migräneanfällen mit und ohne Aura, nach der Erstdiagnose einer Migräne durch einen Arzt, in festen Zubereitungen zur oralen Anwendung in Konzentrationen von 50 mg je abgeteilter Form und in einer Gesamtmenge von 100 mg je Packung“ von der Verschreibungspflicht ausgenommen.
- In der Position „Ibuprofen“ wird die Ausnahme von der Verschreibungspflicht oral anzuwendender flüssiger Zubereitungen für Erwachsene und Kinder von „ab 6 Monaten“ auf „ab 3 Monaten“ geändert.
- Die Positionen „Phenylephrin“, „Fluorescein“ und „Lithium“ werden ergänzt bzw. neu gefasst.
(DAZ 45, S. 112) |



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.