




















- DAZ.online
- DAZ / AZ
- DAZ 7/2015
- Gleich, ähnlich oder ...
Biosimilars
Gleich, ähnlich oder anders?
Wie sich Biosimilars von Originalen unterscheiden können
Seit 2001 begannen nach und nach die Patente einiger wichtiger, umsatzstarker Biopharmazeutika (auch: Biologics, Biologicals, Biologika) auszulaufen. Ab 2006 betraten Nachahmerprodukte, sogenannte Biosimilars, die Bühne. Dies war konsequent und notwendig zur Entlastung des Gesundheitssystems, denn Biopharmazeutika gehören fast ausnahmslos zu den sehr hochpreisigen Arzneimitteln. Die Patentabläufe wichtiger Biopharmazeutika häufen sich zurzeit rasant, sodass in schneller Abfolge mit weiteren Biosimilars gerechnet werden muss (Tab. 1).
| Handelsname | INN | Jahr desPatentablaufs |
|---|---|---|
| Humulin® | Human Insulin | 2001 |
| Cerezyme® | Imiglucerase | 2001 |
| Intron A® | Interferon alfa-2b | 2002 |
| Nutropin®/ - AQ | Somatropin | 2003 |
| Avonex® | Interferon beta-1a | 2003 |
| Humatrope® | Somatropin | 2003 |
| Epogen®/Procrit® | Epoetin alfa | 2004 |
| Synagis® | Palivizumab | 2005 |
| Novolin® | Human Insulin | 2005 |
| Activase® | Alteplase | 2005 |
| Neupogen® | Filgrastim | 2006 |
| Albutein® | Humanalbumin | 2006 |
| Rituxan® | Rituximab | 2013 |
| Erbitux® | Cetuximab | 2014 |
| Remicade® | Infliximab | 2013/2015 |
| Enbrel® | Etanercept | 2015 |
| Herceptin® | Trastuzumab | 2015 |
| Humira® | Adalimumab | 2018 |
| Avastin® | Bevacizumab | 2022 |
Was sind Biosimilars?
In erster Näherung sind Biosimilars „Kopien“ eines seit Jahren bereits zugelassenen Biopharmazeutikums („Referenzarzneimittel“). Da Biopharmazeutika wegen ihrer komplexen Struktur immer eine gewisse molekulare Variabilität aufweisen, kann auch ein Biosimilar strukturell nicht absolut identisch sein mit dem entsprechenden Referenzarzneimittel. Selbst zwischen unterschiedlichen Chargen des Referenzarzneimittels sind Strukturunterschiede nicht vermeidbar. Allerdings sind diese gering und in ihrem Ausmaß strikt limitiert. Dies gilt auch für die minimalen Strukturvariationen zwischen Biosimilar und Referenzarznei, die zudem unter keinen Umständen Auswirkungen auf die Sicherheit oder Wirksamkeit des Arzneimittels haben dürfen. Dies muss durch Daten im Rahmen des zentralen Zulassungsverfahrens bei der EMA (European Medicines Agency) in London nachgewiesen werden und wird dann durch die europaweit erteilte Zulassung auch offiziell bestätigt.
Somit ist ein zugelassenes Biosimilar-Arzneimittel per definitionem genauso wirksam und sicher wie das Referenzarzneimittel. Es wird in derselben Dosis zur Behandlung derselben Krankheiten verwendet wie das Referenzarzneimittel. Und Warnhinweise, die bei der Verabreichung des Referenzarzneimittels zu beachten sind, müssen generell auch beim Einsatz des Biosimilars beachtet werden.
Auf den Punkt gebracht lassen sich Biosimilars wie folgt charakterisieren:
- Ein Biosimilar ist ein Arzneimittel, das einem rekombinanten, bereits zugelassenen Arzneimittel vergleichbar ist (Referenzarznei). Die Wirkstoffe in beiden Arzneimitteln sind ebenfalls ähnlich.
- Biosimilar und Referenzarznei werden in gleicher Dosierung bei gleichen Indikationen eingesetzt.
- Wie für alle anderen Arzneimittel muss auch für Biosimilars eine Zulassung beantragt und erteilt werden, bevor sie verkehrsfähig werden. Die Zulassung wird in Europa durch die Europäische Kommission erteilt, nachdem die EMA als Zulassungsbehörde eine wissenschaftliche Überprüfung der Wirksamkeit, Sicherheit und Qualität des Arzneimittels vorgenommen hat.
- Da der Wirkstoff seit Jahren bekannt ist, müssen nicht alle Informationen eingeholt werden, die bei der Zulassung eines komplett neuen Wirkstoffs erforderlich sind. Verbindliche Richtlinien legen fest, welche Studien durchzuführen sind, um zu belegen, dass das Biosimilar ähnlich und ebenso sicher und wirksam ist wie das Referenzarzneimittel.
- Der Nachweis der strukturellen und funktionellen Übereinstimmung wird durch Vergleichsstudien erbracht. Hierbei handelt es sich um einen „Schritt-für-Schritt-Prozess“, der mit dem Vergleich der Qualität und der Stabilität des Wirkstoffs und des Herstellungsprozesses beginnt. Hierdurch wird belegt, dass keine relevanten Unterschiede hinsichtlich Sicherheit und Wirksamkeit zwischen dem Biosimilar und dem Referenzarzneimittel bestehen.
- Generell folgen die Herstellungsverfahren von Biosimilars denselben Qualitätsstandards wie sie auch für alle anderen neuen Wirkstoffe gelten. Dazu zählt auch, dass die Zulassungsbehörden in bestimmten Abständen die Produktionsstätten inspizieren.
- Da Biosimilar und Referenzarznei ähnlich, aber nicht identisch sind, kann es zumindest bei chronisch kranken Patienten Vorbehalte hinsichtlich der generellen Austauschbarkeit der beiden Arzneimittel geben. Die Entscheidung, einen Patienten mit der Referenzarznei oder mit dem Biosimilar zu behandeln, muss von dem behandelnden Arzt getroffen werden.
- Alle Arzneimittel – also auch Biosimilars – werden nach ihrer Zulassung hinsichtlich ihrer Sicherheitsprofile beobachtet.
- Hierzu muss jeder pharmazeutische Hersteller ein Pharmakovigilanz-System etablieren, das jede Art von Auffälligkeiten – besonders auch immunologische Auffälligkeit – registriert. Dieses System wird ebenfalls von der Zulassungsbehörde überprüft. Sollte es Anlass zu besonderer Vorsicht geben, muss das Biosimilar die gleichen Auflagen erfüllen wie das Referenzarzneimittel. Hierzu gehört beispielsweise die Erstellung eines besonderen Risk-Management-Plans.
Die Zulassung von Biosimilars
Alle gentechnisch hergestellten Arzneimittel müssen in einem zentralisierten Verfahren über die EMA zugelassen werden. Für die Zulassung von Biosimilars musste ein eigenes gesetzliches Regelwerk entwickelt werden. Seit 2003 wurde bei der EMA an diesem Zulassungsprozess gearbeitet. Die Basis bildete die Direktive 2001/83/EC, die umfassend überarbeitet wurde. Zusätzlich wurde eine ganze Reihe von regulatorischen Leitlinien erstellt, in denen die Details für das Zulassungsverfahren beschrieben werden (Abb. 1).
Abb. 1: Richtlinien mit Relevanz für die Zulassung von Biosimilars durch die EMA. EPO: Erythropoetin, FSH: Follikel-stimulierendes Hormon, G-CSF: Granulozyten-Kolonie-stimulierender Faktor, IFN: Interferon, LMWH: niedermolekulares Heparin, MAB: monoklonaler Antikörper.
Seit 2005 sind die gesetzlichen Bestimmungen für die Zulassung von Biosimilars in der EU in Kraft. Damit hat die EMA auf dem Gebiet der Zulassung von Biosimilars weltweit die Vorreiterrolle übernommen. Als Resultat dieser regulatorischen Pionierarbeit stehen heute qualitativ hochwertige Nachfolgepräparate von ehemals innovativen Biopharmazeutika zur Verfügung, die nach Bestehen dieses Zulassungsverfahrens als Biosimilars vermarktbar sind. Nur solche Kopien von biopharmazeutischen Nachahmerprodukten, die dieses Zulassungsverfahren erfolgreich durchlaufen haben, sind als Biosimilars in Europa verkehrsfähig. Präparate ohne EMA-Zulassung sind in Europa nicht verkehrsfähig. Sie verdienen nicht das Qualitätsprädikat „Biosimilar“, denn sie zeigen zum Teil gefährliche Qualitätsmängel.
Nicht jeder zur Zulassung bei der EMA eingereichte Biosimilar-Kandidat erfüllt die hohen Ansprüche, die an diese Arzneimittel gestellt werden. Dies unterstreicht die Gleichwertigkeit von Biosimilars mit den entsprechenden Referenzprodukten nicht nur hinsichtlich ihrer Wirksamkeit, sondern auch hinsichtlich ihrer Qualität und Unbedenklichkeit. Zugleich unterstreicht dies, wie wichtig es ist, dass nur in der EU zugelassene Biosimilars therapeutisch eingesetzt werden dürfen.
| Handelsname, Hersteller | INN | Referenzprodukt | Entscheidung | Datum der Entscheidung |
|---|---|---|---|---|
| Omnitrope®, Sandoz | Somatropin | Genotropin® | zugelassen | 12. 4. 2006 |
| Valtropin®, BioPartners | Somatropin | Humatrope® | vom Markt genommen | 24. 4. 2006 |
| Biferonex®, BioPartners | Interferon beta-1a | Avonex® | zurückgezogen | 28. 5. 2009 |
| Alpheon®, BioPartners | Interferon alfa-2a | Roferon-A® | abgelehnt | 5. 9. 2006 |
| Binocrit®, SandozEpoetin alfa Hexal®, HexalAbseamed®, Medice | Epoetin alfa | Eprex® | zugelassenzugelassen zugelassen | 28. 8. 2007 |
| Retacrit®, HospiraSilapo®, Stada | Epoetin zeta | Eprex® | zugelassenzugelassen | 18. 12. 2007 |
| Epostim®, Reliance GeneMedix | Epoetin alfa | Eprex® | zurückgezogen | 15. 3. 2011 |
| Insulin Human Rapid / Long /30/70 Mix Marvel®, Marvel | Insulin | Humulin® | zurückgezogen | 20. 12. 2007 |
| Solumarv®, MarvelIsomarv®, MarvelCombimarv®, Marvel | Insulin | Humulin® | zurückgezogenzurückgezogenzurückgezogen | 15. 11. 2012 |
| Biograstim®, CT ArzneimittelFilgrastim ratiopharm®, RatiopharmRatiograstim®, RatiopharmTevagrastim®, Teva Generics | Filgrastim | Neupogen® | zugelassenvom Markt genommenzugelassenzugelassen | 15. 9. 2008 |
| Zarzio®, SandozFilgrastim Hexal®, Hexal | Filgrastim | Neupogen® | zugelassenzugelassen | 6. 2. 2009 |
| Grastofil®, Stada | Filgrastim | Neupogen® | zugelassen | 18. 10. 2013 |
| Nivestim®, Hospira | Filgrastim | Neupogen® | zugelassen | 8. 6. 2010 |
| Inflectra®, HospiraRemsima®, Celltrion | Infliximab | Remicade® | zugelassenzugelassen | 10. 9. 2013 |
| Ovaleap®, Teva | Follitropin alfa | GONAL-f® | zugelassen | 27. 9. 2013 |
| Bemfola®, Finox Biotech | Follitropin alfa | GONAL-f® | zugelassen | 27. 3. 2014 |
| Basaglar® , Lilly u. Boehringer Ingelheim | Insulin glargin | Lantus® | zugelassen | 10. 9. 2014 |
Die Tabelle 2 fasst die bisherigen zentralen Zulassungsverfahren für Biosimilars bei der EMA zusammen. Zum Beispiel listet die EMA-Homepage* einen Interferon-alfa-2a-Wirkstoff, dem die Zulassung 2006 versagt wurde. Ende 2007 wurden die Zulassungsanträge für drei Insulin-Biosimilars zurückgezogen, da das Unternehmen die ihm vom Ausschuss für Humanarzneimittel der EMA (Committee for Medicinal Products for Human Use, CHMP) gestellten Fragen zur Herstellung und zur Wirksamkeit nicht innerhalb der vorgegebenen Frist beantworten konnte. Im März 2011 wurde der Zulassungsantrag für ein Epoetin-Biosimilar zurückgezogen, da das Unternehmen die Fragen des CHMP nicht fristgerecht beantworten konnte. Und schließlich wurden im November 2012 die Neueinreichungen von drei Zulassungsanträgen für Insulin-Biosimilars (s. o.) zurückgezogen, nachdem die EMA Bedenken mit Blick auf die Herstellung der Biosimilars sowie die Qualität der zugrunde liegenden klinischen Daten geäußert hatte.
Regulatorische Situation außerhalb der EU
Nicht nur in Europa gibt es ein Regelwerk für die Zulassung von Nachfolgeprodukten von patentfreien Biopharmazeutika. Allerdings sind die Standards bisher keinesfalls harmonisiert, weshalb eine gegenseitige Anerkennung auch nicht möglich ist.
Die EMA kooperiert mit folgenden Partnern:
- Health Canada (Eine finalisierte Richtlinie „Guidance on Subsequent Entry Biologics“ wurde im März 2010 publiziert.)
- Japan (Eine Richtlinie „Guideline on quality, safety and efficacy of follow-on biologics“ wurde im März 2009 publiziert.)
- WHO (Eine Richtlinie „Guidelines on Evaluation of Similar Biotherapeutic Products“ wurde im Oktober 2009 verabschiedet.)
- FDA (Die Erarbeitung eines verkürzten Zulassungsverfahrens für Biosimilars im Rahmen des Patient Protection and Affordable Care Act wurde am 23. März 2010 beschlossen, aber noch nicht abgeschlossen.)
Was muss ein Biosimilar nachweisen, bevor es von der EMA zugelassen wird?
Um die Zulassung für ein Biosimilar zu erhalten, muss der Hersteller die Qualität, Sicherheit und Wirksamkeit im Vergleich zur Referenzarznei belegen. Das Verfahren, das als „comparability exercise“ bezeichnet wird, ist sequenziell ausgelegt und erfolgt in drei Schritten.
Im ersten Schritt werden die Vergleichbarkeit der Qualität bzw. die physikalisch-chemische und die biologische Vergleichbarkeit demonstriert. Hier wird die strukturelle Übereinstimmung von Biosimilar und Referenzarznei in allen relevanten Details mit einem riesigen Spektrum an analytischen Methoden belegt. Mögliche Abweichungen von den Daten der Referenzarznei müssen plausibel erklärt werden. Ferner wird die Reinheit des Biosimilars überprüft, und das Produkt wird nur freigegeben, wenn die im Vorfeld definierten Spezifikationskriterien erfüllt werden.
Im zweiten Schritt werden Biosimilar und Referenzarznei im präklinischen Setting miteinander verglichen. In aller Regel kann hier auf ein verkürztes Verfahren in Form von In-vitro-Untersuchungen zugegriffen werden. Diese Untersuchungen sind in produktspezifischen Richtlinien durch die EMA vorgegeben. Die Pharmakokinetik/Pharmakodynamik-Parameter und deren vordefinierter Grad der Ähnlichkeit mit der Referenzarznei müssen begründet und getroffen werden.
Im dritten Schritt wird dann die klinische Vergleichbarkeit belegt. Diese Studien haben mehr den Charakter von Sicherheitsstudien als von Wirksamkeitsstudien. Denn wenn im ersten Schritt des „comparability exercise“ belegt worden ist, dass Biosimilar und Referenzarznei ausreichend ähnlich sind, dann ist auch damit zu rechnen, dass sie klinisch äquivalent wirken. Zwingend sind diese klinischen Studien jedoch gefordert, um die Verträglichkeit der Biosimilars zu belegen. Denn die Herstellungsprozesse von Biosimilar und Referenzarznei sind zwangsläufig unterschiedlich, sodass nicht ausgeschlossen werden kann, dass analytisch nicht oder nur schwer fassbare Komponenten eine klinische Auffälligkeit provozieren. In klinischen Phase-I-Studien liegt der Fokus zunächst auf der Toxikologie, der Pharmakokinetik und der Pharmakodynamik. Das heißt, der Wirkstoff wird auf seine Reinheit und Unbedenklichkeit hin geprüft, seine Verarbeitung im Körper nachvollzogen (Aufnahme, Verteilung im Körper, biochemischer Auf- und Umbau sowie Ausscheidung) und ein Wirkprofil erstellt. Daran schließen sich Studien zur Wirksamkeit und Sicherheit im Sinne von Schwere und Häufigkeit verschiedener Nebenwirkungen bei einer oder mehreren repräsentativen Indikationen an, um ein vergleichbares Wirksamkeits- und Sicherheitsprofil zu demonstrieren. Hierzu zählt auch ein vergleichbares Immunogenitätsprofil von Biosimilar und Referenzarznei. Schwerpunkte und Anforderungen an diese Phase-III-Studien sind je nach Biosimilar-Klasse verschieden. Entsprechend der Unterschiedlichkeit und Komplexität von Biopharmazeutika legt die EMA die Anforderungen individuell und an den Leitlinien orientiert mit den Herstellern fest.
„Ähnlich“ statt „identisch“: Wie „gleich“ sind Biosimilars?
Die enorme strukturelle Komplexität von Proteinen lässt es nicht zu, dass eine Präparation eines bestimmten Proteins in absolut reiner Form vorliegt. So enthält eine Proteinlösung immer auch strukturell leicht unterschiedliche Molekülformen, die dadurch entstehen, dass einzelne Aminosäuren durch chemische Reaktionen (Deamidierung oder Oxidation) verändert wurden, dass an den Enden Aminosäuren abgeschnitten wurden, dass sich die Tertiärstruktur in Teilen verändert hat oder dass sich Aggregate gebildet haben.
Alle diese Modifikationen, die letztlich nur einen sehr kleinen Teil der Proteinpräparation ausmachen, lassen sich heute analytisch nachweisen. Da sie de facto unvermeidlich sind, müssen und können sie auch akzeptiert werden, vorausgesetzt, sie verursachen keine auffälligen Reaktionen, wenn die Proteinpräparation beim Menschen therapeutisch eingesetzt wird.
Um hier Sicherheit zu garantieren, werden die Bedingungen der Herstellung und Lagerung von Biopharmazeutika extrem konstant gehalten („The Product is the Process“). Dies wiederum garantiert auch eine gewisse Konstanz der Heterogenität der Präparationen, die durch Spezifikationsgrenzen nach oben und nach unten definiert sind. Innerhalb dieser Spezifikationsgrenzen haben sich heute auch Biosimilars zu bewegen. Man kann daher in erster Näherung konstatieren, dass Biosimilars in dem Maße der Referenzarznei ähneln, wie sich einzelne Chargen der Referenzarznei untereinander ähneln.
Extrapolation der Indikationen
Ist das Referenzprodukt für mehr als eine Indikation zugelassen, erlaubt die EMA, basierend auf einer Fall-zu-Fall-Bewertung, für das Biosimilar die Extrapolation auf alle zugelassenen Indikationen der Referenzarznei, obwohl das Biosimilar nur in einer Indikation klinisch getestet wurde. Voraussetzung ist allerdings, dass den unterschiedlichen Indikationen der gleiche Wirkmechanismus des Wirkstoffs zugrunde liegt und es keine wissenschaftlichen Einwände gibt.
Diese Besonderheit im Zulassungsprozess von Biosimilars stößt bei Klinikern auf sehr große Skepsis. Für Pharmazeuten ist diese Entscheidung wesentlich besser nachvollziehbar, da Pharmazeuten ausbildungsbedingt ein besseres molekulares Verständnis besitzen. Denn wenn sichergestellt ist, dass Referenzarznei und Biosimilar ausreichend ähnlich sind, ist es absolut plausibel, dass das Biosimilar auch in allen Indikationen wirksam und sicher ist, für die die Referenzarznei zugelassen ist.
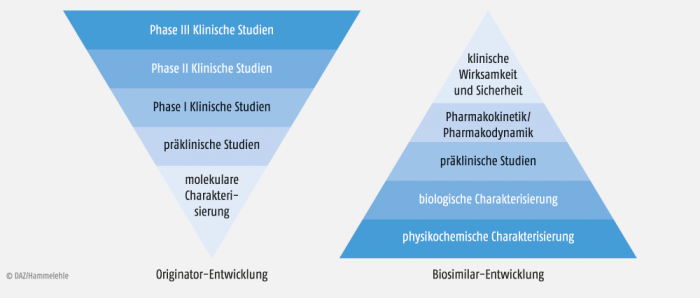
Biosimilars werden hingegen „umgekehrt“ entwickelt (reversed engineering). Hier steht im Vordergrund der Nachweis der möglichst exakten Kopie der Referenzarznei. Wirksamkeit und Sicherheit ergeben sich dann automatisch und werden auf Basis der nachgewiesenen strukturellen Ähnlichkeit in relativ kleinen klinischen Studien nur noch bestätigt.
Das Prinzip der Entwicklung eines Biosimilars beruht auf einem Reversed-engeneering-Ansatz, wobei die Evidenz im Labor generiert wird, die in der Klinik in relativ kleinen Studien nur überprüft wird (Abb. 2). Dies ist kein „fauler Kompromiss“, wie vielfach geargwöhnt wird. Vielmehr sind Labortests um ein vielfaches sensitiver als klinische Tests, wo kleine Unterschiede kaum auffallen.
Pharmakovigilanz von Biosimilars
Die Überwachung eines Arzneimittels nach seiner Markteinführung mit Blick auf Sicherheit und Nebenwirkungen gehört zur Gewährleistung der generellen Arzneimittelsicherheit und gilt natürlich auch für Biosimilars.
Besondere Aufmerksamkeit verdienen dabei unerwünschte immunologische Reaktionen. Hier liegt keine besondere Gefahr bei den Biosimilars. Vielmehr muss bei Biopharmazeutika generell ein besonderes Augenmerk auf immunologische Unverträglichkeiten gelegt werden. Dies liegt zum einen in der makromolekularen Struktur begründet. Zum anderen können minimale Verunreinigungen während der Produktion, die aus der Wirtszelle oder dem Fermentationsmedium stammen können, beim Patienten eine immunologische Reaktion provozieren.
Die Erfahrung hat aber gezeigt, dass dieses Problem in der Praxis viel kleiner ist, als es ursprünglich antizipiert wurde. Biosimilars, so die Erfahrung aus millionenfachem Einsatz, sind ebenso sicher einzusetzen wie die entsprechende Referenzarznei.
Monoklonale Antikörper (MAB) …
… werden das Spektrum der Biosimilars noch einmal erweitern. Das erste MAB-Biosimilar (Inflectra®/Remsima®) mit dem Wirkstoff Infliximab wurde im September 2013 in Europa zugelassen. Es handelt sich bei beiden Präparaten um sogenannte Bioidenticals, die dem gleichen Herstellungsprozess entstammen und von der südkoreanischen Firma Celltrion Healthcare (Remsima®) bzw. von der US-amerikanischen Firma Hospira (Inflectra®) vermarktet werden. In Deutschland wird Remsima® von Mundipharma vertrieben werden. Als Referenzarznei für die Entwicklung diente Remicade® der Firma MSD, das wie seine Biosimilars für folgende Indikationen zugelassen ist: rheumatoide Arthritis, ankylosierende Spondylitis, Morbus Crohn, Colitis ulcerosa, Psoriasis-Arthritis und Psoriasis.
Kurz vor der Zulassung dieses ersten Biosimilar-Antikörpers konnte die Firma Janssen Biotech Inc. eine Verlängerung ihres Patents für Remicade® bis Anfang 2015 erwirken, sodass die Einführung von Inflectra®/Remsima® erst Ende Februar erfolgen kann. Damit ist ein wichtiger Anfang gemacht. Das lange beschworene „Dogma“, rekombinante, monoklonale Antikörper ließen sich nicht kopieren, ist seit der Erteilung der Zulassung des Infliximab-Biosimilars nicht mehr zu halten.
Weitere monoklonale Antikörper sind als Biosimilars in der fortgeschrittenen Entwicklung, und mit deren Markteinführung muss gerechnet werden, sowie die Patente der Referenzprodukte auslaufen. |
Autoren
Prof. Dr. Theo Dingermannist Seniorprofessor am Institut für Pharmazeutische Biologie an der Goethe-Universität Frankfurt.
Dr. Ilse Zündorf ist dort als akademische Oberrätin tätig.
Institut für Pharmazeutische Biologie
Biozentrum
Max-von-Laue-Straße 9
60438 Frankfurt/Main
Foto: nbiebach - Fotolia.com



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.